Resumen paiper- Gabriela Londoño Torres
HEMOSTASIA
constituye el conjunto de mecanismos fisiológicos, moleculares y celulares, dirigidos a evitar
la pérdida de sangre o hemorragia, debida a la rutura de vasos sanguíneos o del sistema
conductor de la sangre. Los sistemas de la hemostasia requieren la interacción de tres
estructuras : La pared del endotelio vascular, las plaquetas y un conjunto de proteínas y
otros elementos plasmáticos que se agrupan bajo el nombre de factores de la coagulación .
El estímulo que desencadenará la activación de la hemostasia es la lesión a nivel del
endotelio provocando el contacto de la sangre con el tejido conectivo subendotelial.
La hemostasia consta de 3 componentes principales cuyos efectos se superponen
parcialmente en el tiempo e interactúan entre sí.
● Hemostasia primaria : Se activa en los primeros minutos de una hemorragia.
Depende principalmente de los vasos y de las plaquetas. (da lugar a la formación del
trombo plaquetario)
● Hemostasia secundaria: o formación del coágulo y reparación de los tejidos dañados
● Fibrinólisis : o destrucción enzimática del coágulo, restableciendo la circulación una
vez que este ha cumplido su función .
Existe siempre una interacción entre la pared vascular y la sangre. Los fenómenos de la
hemostasia que ocurren sucesivamente en el lugar de la lesión vascular se pueden resumir
de la siguiente manera : Después de una lesión inicial, se produce una vasoconstricción
arteriolar de breve duración, que en gran parte se distribuye a mecanismos neurógenos
reflejos y que se acentúa con la secreción local de ciertos factores, como la endotelina
(potente vasoconstrictor derivado del endotelio). La lesión del endotelio deja al descubierto
la matriz extracelular subendotelial (con gran poder trombogénico) que permite a las
plaquetas adherirse y activarse, es decir, sufrir un cambio de forma y vaciar sus
granulaciones secretoras, en pocos minutos atraen a otras plaquetas y se forma el tapón
hemostático (hemostasia primaria ). El factor tisular actúa junto a los factores secretados
por las plaquetas para activar la cascada de coagulación y culmina con la activación de la
trombina. Ésta a su vez convierte al fibrinógeno disuelto en la sangre en fibrina insoluble,
que termina depositando localmente; esta serie de eventos conforman la hemostasia
secundaria y dura más tiempo que la formación del tapón plaquetario.
La fibrina polimerizada y los agregados de plaquetas forman un sólido tapón permanente,
que impide cualquier nueva hemorragia. En Esta fase se ponen en marcha los mecanismos
de contrarregulación para que el tapón quede circunscrito al sitio de la lesión, por ejemplo,
el activador del plasminógeno tisular (t -PA)

HEMOSTASIA PRIMARIA.
Vasoconstricción : Cuando la pared de un vaso se lesiona, su calibre tiende a reducirse por
intensa contracción del músculo liso de la propia pared vascular que reduce el flujo de
sangre. El flujo puede interrumpirse por este mecanismo en vasos pequeños como
arteriolas y vénulas. Los capilares carecen de músculo liso, pero en ellos el flujo puede
detenerse por contracción de los esfínteres pre-capilar.
La vasoconstricción se produce por un triple mecanismo:
1. Neurogénico : Dependiente de la inervación autonómica del vaso.
Es mediado por noradrenalina liberada por las fibras postganglionares simpáticas.
2. Miogénico: Independiente de la inervación.
Se debe a la respuesta del músculo liso a la lesión, y a la falta de liberación de
sustancias vasodilatadoras ( como óxido nítrico y prostaciclina) por el endotelio
ausente en la zona de la lesión .
3. Hemogénico; Debido a sustancias vasoconstrictoras liberadas por las plaquetas
(serotonina, tromboxano A2). Es más duradero que los anteriores.
FORMACIÓN DEL TAPÓN PLAQUETARIO:
Las plaquetas o trombocitos son células discoides anucleadas, en su citoplasma contienen
concentración elevada de calcio, en su mb hay microtúbulos además de la proteína
contráctil trombostenina formada por actina y miosina.
Las plaquetas tienen un sistema canalicular continuo con el LEC que penetra al interior de
ellas. La mb plasmática está recubierta por un denso glicocálix, en el cual hay varias
glicoproteínas integrales de mb que funcionarán como receptores y transductores de
señales intracelulares necesarios para la activación de las plaquetas.
Los trombocitos poseen tres clases de gránulos:
1.Gránulos alfa : Contienen fibrinógeno, factor de crecimiento derivado de
plaquetas(PDGF), factor de von willebrand, P-selectina y otras proteínas
2. Gránulos delta : ADP, ATP Y 5-hidroxitriptamina
3. Gránulos lambda: son lisosomas con enzimas proteolíticas.
La formación del tapón plaquetario incluye fenómenos que pueden resumirse así :
● Las plaquetas se adhieren a la matriz extracelular en los sitios lesionados del
endotelio y se activan
● Una vez activadas , secretan los productos que contienen sus granulaciones y
sintetizan TXA2.
● Exponen complejos de fosfolípidos que son importantes para la vía intrínseca de la
coagulación.
● Las células endoteliales lesionadas o activadas se exponen al factor tisular , que
desencadena la cascada extrínseca de la coagulación .

● La ADP liberado por las plaquetas favorece la formación de un tapón hemostático
primario, que luego se convierte en un tapón secundario más grande y definitivo.
● El depósito de fibrina actúa estabilizando y sirviendo de anclaje a las plaquetas
agregadas.
Adhesión plaquetaria :
Las plaquetas circulantes se dirigen hacia la pared del vaso, lo cual es facilitado porque los
eritrocitos tienden a circular por el centro del vaso y marginan a las plaquetas. Normalmente
las plaquetas circundantes no se adhieren entre sí ni al endotelio intacto.
La adhesión plaquetaria se debe a la lesión del endotelio, que expone el colágeno de la
matriz extracelular perivascular. El factor de von willebrand se sintetiza en el endotelio y se
almacena en los cuerpos de weibel-palade, desde donde es secretado de manera
constitutiva y también frente a estímulos. como se dijo antes, los gránulos alfa de las
plaquetas también transportan factor de Von willebrand pero no lo liberan constitutivamente,
sino sólo en respuesta a estímulos
Ante una lesión vascular que daña el endotelio, el factor de Von willebrand se adhiere al
colágeno y sirve como un adaptador para la adhesión de las plaquetas. En ausencia del
factor de von willebrand , la adhesión de los trombocitos al colágeno es difícil y fácilmente
desecha por el propio flujo sanguíneo.
Activación :
Tras el contacto de la plaqueta con el colágeno o bien con otra plaqueta, éste se activa
produciendo alteraciones morfológicas y bioquímicas que requieren de energía.
Por un lado adquieren forma más esférica y emiten seudópodos. De esta forma se aumenta
la superficie de contacto favoreciendo la unión de unas con otras. Se produce liberación del
contenido de los gránulos plaquetarios, Secretando calcio, ADP, fibrinógeno, serotonina y
factor plaquetario 4 con capacidad de reclutas más plaquetas y aumentar su actividad
agregante, por otro lado se exponen los fosfolípidos de l mb plaquetaria que son conocidos
como F3P (factor plaquetario 3 ) que ponen en marcha una reacción autocatalítica, que
conduce a la formación de un agregado creciente de plaquetas, al tapón hemostático
primario.
Ésta agregación es reversible, pero al activar la cascada de la coagulación, se forma
trombina, la cual se une al receptor de superficie de las plaquetas, que junto con el ADP y el
TXA2 producen más agregación. El TXA2 favorece la agregación plaquetaria estimulando la
desgranulación de la plaqueta. (el TXA2 se forma a partir del ácido araquidónico)
La prostaciclina ( formada a partir de ácido araquidónico también) se forma en el endotelio
vascular y tienen un efecto antagónico al del TXA2 , tienen acción vasodilatadora y
antiagregante.
El equilibrio entre PGI2 y TXA2 ,Limita la formación de trombos plaquetarios .

HEMOSTASIA SECUNDARIA-COAGULACIÓN.
La coagulación se debe a la transformación del fibrinógeno en fibrina y la polimerización de
esta para formar una malla insoluble.
Participan proteasas de serina presentes en la sangre como zimógenos.
Pueden distinguirse 3 grupos de factores de la coagulación.
1. Factores dependientes de la vitamina K:
Son protrombina y los factores VII, IX, X. En los pasos de la cascada que requiere
Calcio (Ca+2) este ión participa permitiendo la unión de la proteasa conato y el
cofactor proteínico.
2. Factores sensibles a la trombina :
Incluyen al fibrinógeno y a los factores V, VII, VIII, Y XII. son de alto peso molecular y
se consumen en el proceso de coagulación. Los factores V, VIII y el factor tisular, si
bien no son enzimas, aceleran las reacciones enzima-sustrato en las que
intervienen.
3. Factores de contacto : Factores XII, XI, La precalicreína (factor Fletcher) y el
Kininógeno de alto peso molecular. Todos actúan en la primera fase de la
coagulación y también desencadenan la fibrinólisis.
INHIBIDORES NATURALES DE LA COAGULACIÓN SANGUÍNEA.
Existen 3 tipos principales.
● Inhibidores de las serinproteasas : antitrombina III,es el principal inhibidor de la
trombina , la heparina ; es un anticoagulante producido por las células cebadas y los
basófilos , que en combinación con AT-II aumentan la eficacia de esta en el bloqueo
de la trombina, alfa 2 macroglobulina, contribuye un 25% del total de la actividad
antitrombina del plasma.
también inhiben la calicreína y la plasmina.
● Inhibidores de los factores VIIIa y Va: Proteína C, que es una serinproteasa que es
activada por la trombina para ejercer su acción anticoagulante. Proteína S, es una
glicoproteína dependiente del calcio que funciona como cofactor de la proteína C en
la inactivación de los factores Va y VIIIa.
● Inhibidor de la vía del factor tisular:Este inhibidor está en su mayoría unido a la
fracción LDL en el plasma . Se lo ha denominado anti tromboplastina, inhibidor
tisular (TFPI), inhibidor de la vía extrínseca (EPI).

FIBRINOLISIS
Es el proceso por el cual se elimina el coágulo de fibrina una vez que se ha restaurado la
integridad de la estructura vascular , el cual se desarrolla a través del sistema
plasminógeno-plasmina.
La base de la fibrinolisis es la transformación de una proenzima plasmática inactiva, el
plasminógeno, en una enzima activa, la plasmina; con alta actividad proteolítica y capacidad
para digerir los anillos de fibrina, el fibrinógeno y desactivar otros factores de la coagulación
(V y VIII)
Los activadores del plasminógeno se pueden dividir en dos categorías:
● Activadores intrínsecos: incluyen el factor XIIa, la calicreína y el factor XI a.
● Activadores extrínsecos: están distribuidos en todos los tejidos del organismo y son
sintetizados por el endotelio vascular que ante determinados estímulos, como
endotoxinas bacterianas y el factor de necrosis tumoral los libera a la circulación.
se denomina activador tisular del plasminógeno(t-PA).
Como consecuencia de la acción de estos factores, el plasminógeno se transforma en
plasmina; la plasmina se encarga de romper la fibrina del coagulo, el fibrinógeno, el factor V
y el factor VIII , convirtiéndolos en productos inactivos o de degradación.
La plasmina posee una actividad muy intensa, por lo que se debe controlar para que no
ocurran respuestas anticoagulantes desmesuradas. Para ello existen dos sistemas de
control:
● La actividad de la alfa2 -antiplasmina circulante (principal inhibidor de la plasmina)
● La existencia de sustancias capaces de inhibir la actividad del t-PA (plasminógeno
activador inhibidor, PAI)
La regulación de la fibrinolisis se produce por :
1. liberación de un activador plasminógeno por el endotelio vascular activado o
lesionado , llamado activador tisular (t-PA)
2. la depuración hepática del activador del plasminógeno
3. la activación del plasminógeno
4. la inhibición de la activación del plasminógeno (PAI-1)
La aspirina y el clopidogrel son fármacos que inhiben la agregación plaquetaria. Los
quelantes de calcio se emplean como anticoagulantes de uso in vitro. La heparina tiene
actividad anticoagulante tanto in vitro como in vivo. Los antagonistas de la vitamina K.
Pruebas de laboratorio para Detectar trastornos vasculares y de las plaquetas
Prueba del torniquete: Se realiza aplicando un manguito de presión en el brazo, se
mantiene 5 min a un nivel situado entre las presiones sistólicas y diastólica; se examina
entonces por debajo del manguito para observar si se ha producido o no petequias(
pequeñas manchas rojas de tamaño aprox al de la cabeza de un alfiler) .
Tiempo de sangría : Mide el tiempo que tarda en detenerse la hemorragia luego de realizar
una incisión en el lóbulo de la oreja. Esta prueba mide las primeras etapas de la formación
del tapón hemostático. El tiempo de sangría se prolonga cuando existen alteraciones de las
plaquetas cualitativas o cuantitativas , déficit del factor de von willebrand o ingestión de
fármacos antiagregantes plaquetarios como la aspirina. VN : 1-4 min
Recuento de plaquetas : el número normal de plaquetas en sangre es entre 150.000 y
400.000/mm3. Detecta trombocitopenia o trombocitosis.
Agregación plaquetaria in vitro : miden la respuesta de los trombocitos a diferentes
agonistas como ADP, ácido araquidónico ( se transforma en tromboxano A2) , colágeno o
adrenalina.
Pruebas para detectar trastornos en el proceso de coagulación
Tiempo de coagulación de sangre total-método de Lee-White:
Se observa la formación del coágulo en tubos de vidrio en condiciones estandarizadas, esta
prueba mide el mecanismo intrínseco de la coagulación.
El tiempo normal de coagulación en tubo es de 5-11 min a 37°C. El tiempo de coagulación
está prolongado cuando hay severa deficiencia de todos los factores de coagulación.
Tiempo de tromboplastina parcial activada (KPTT): Se mide el tiempo que demora en
formarse un coágulo de fibrina en plasma citratado al cual se le agrega calcio, fosfolípidos y
partículas que actúan como factor de contacto como el caolín.
El KPTT se prolonga en las deficiencias de factores de la vía intrínseca y común.
Tiempo de trombina (TT): mide el tiempo durante el cual el fibrinógeno presente en el
plasma se transforma en fibrina por la adición de una cantidad estandarizada de trombina.
Son causas comunes de prolongación del tiempo de trombina; presencia de heparina o
aumento de los productos de degradación del fibrinógeno/fibrina.
Tiempo de protrombina: Es el tiempo que tarda en formarse el coágulo de fibrina en plasma
citratado luego de agregar calcio y tromboplastina.( un extracto de factor tisular y
fosfolípidos).
se prolonga en la deficiencia de factor VII, de algún factor de la vía final común
Concentración de fibrinógeno: La concentración normal es de 150 a 400 mg/dL. puede
estar bajo por afibrinogenemia o por consumo excesivo.

Los productos de degradación de la fibrina, en particular los dímeros D de fibrina, aumentan
en plasma como resultado de la fibrinolisis.
La coagulación IN-VITRO
Cuando se extrae sangre y se coloca en un tubo de ensayo, la coagulación se produce en 6
a 10 min. Esto puede evitarse si se le reduce la concentración de Calcio , por ejemplo con
citrato de sodio. En estas condiciones la sangre permanece sin coagular por tiempo
indefinido.
La vía intrínseca, es iniciada por el contacto con una superficie extraña, como vidrio o los
cristales de caolín.
La vía extrínseca, se inicia cuando el factor tisular (factor III), una glicoproteína presente en
los tejidos, se une al factor VII presente en el plasma.
Ambas vías convergen en la activación del factor X.
El factor Xa, con calcio, fosfolípidos Va como cofactores ,Cataliza la transformación de la
protrombina (Factor II) en trombina (IIa). La trombina escinde dos péptidos del fibrinógeno
(factor I), transformándolo en fibrina.
La coagulación IN-VIVO
Las vías intrínseca y extrínseca no pueden funcionar fisiológicamente como vías
alternativas y redundantes, sino que son complementarias. Los fosfolípidos que se añaden
en los ensayos in vitro, como el TTPK, son proporcionados in vivo por superficies celulares,
donde participan dos tipos de células : las que expresan en su superficie el factor tisular y
las plaquetas.
Fase de iniciación : Cuando se produce extravasación de la sangre, el factor VII plasmático
se une al factor tisular en la superficie de las células.(la unión al factor tisular aumenta la
actividad proteolítica del factor VII).
Fase de amplificación:
Las pequeñas cantidades de trombina generadas en la superficie de las células que
expresan factor tisular cumplen funciones diversas: 1. actuar como agonista para la
activación plaquetaria , 2 activar los cofactores V y VIII sobre la superficie plaquetaria
3.Activar al zimógeno factor XI en el mismo lugar.
Todas estas acciones posibilitan una generación mayor de trombina durante la fase de
amplificación y promocionan las condiciones para la fase siguiente.
Fase de propagación :
Esta 3ra fase acontece sobre plaquetas activadas. En superficie se produce una generación
de factor Xa suficiente para desencadenar un pico de generación de trombina suficiente
para escindir la cantidad de fibrinógeno necesaria para que se forme la red de fibrina.
Una vez formado el tapón hemostático madura, la pared del vaso debe ser reparada. La
trombina tiene un papel importante en reclutar y activar las células involucradas en la
reparación, como macrofagos, fibroblastos, células endoteliales y células musculares.
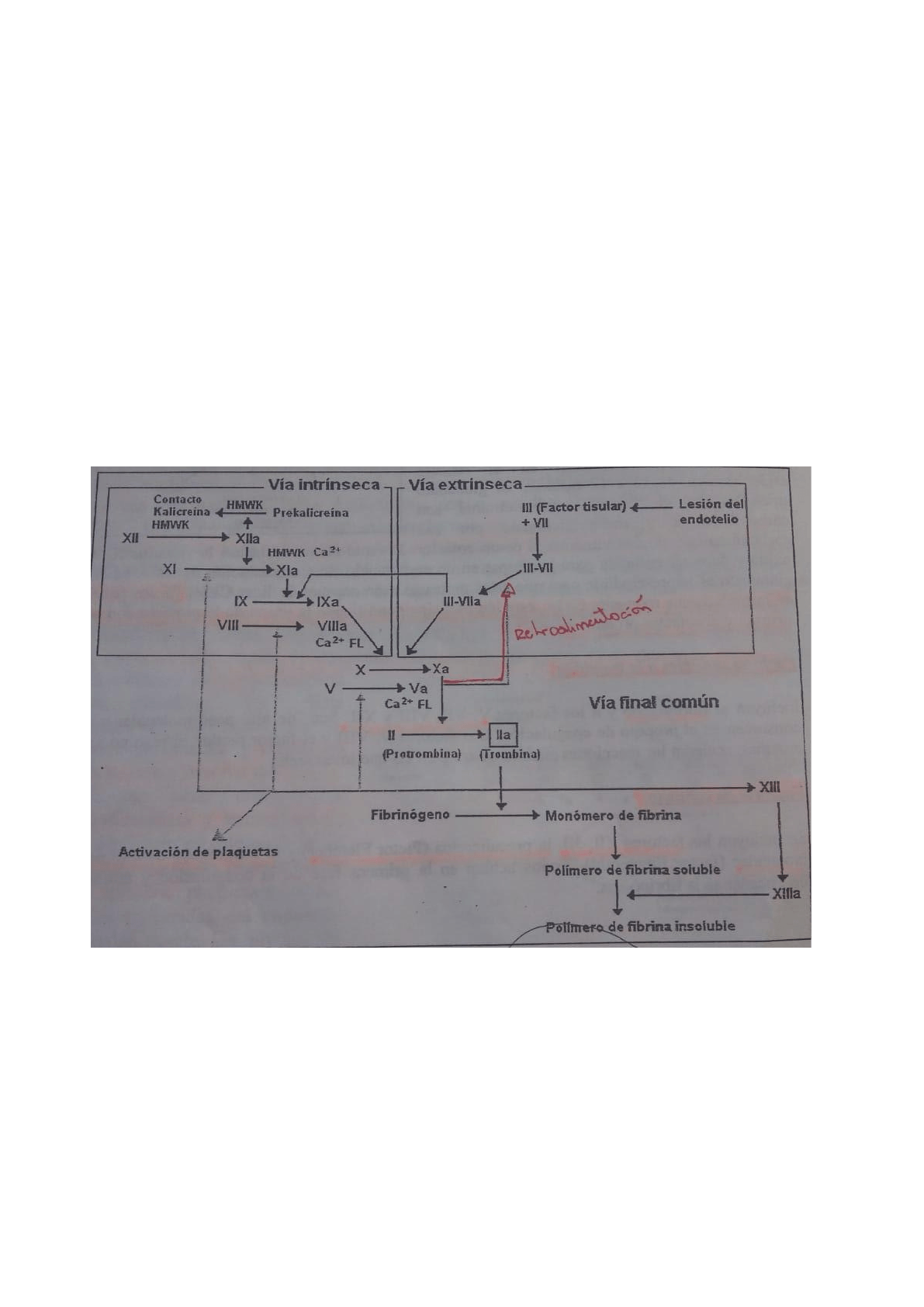
En resumen, la coagulación In vivo es iniciada sobre células que expresan un factor tisular y
clásicamente pertenecen a la llamada vía extrínseca. El proceso es transferido a la
superficie de las plaquetas durante la fase de amplificación y en ellas tiene lugar también la
generación de trombina a gran escala , principalmente mediada por componentes de la vía
intrínseca.
La tasa de producción de trombina y la concentración de trombina una vez formada la red
de fibrina contribuyen a determinar las propiedades del coágulo.

RESUMEN -HEMOSTASIA.pdf
 Estamos procesando este archivo...
Estamos procesando este archivo...
 Lamentablemente la previsualización de este archivo no está disponible. De todas maneras puedes descargarlo y ver si te es útil.
Lamentablemente la previsualización de este archivo no está disponible. De todas maneras puedes descargarlo y ver si te es útil.
Descargar
Estamos procesando este archivo...
Lamentablemente la previsualización de este archivo no está disponible. De todas maneras puedes descargarlo y ver si te es útil.