
Revista de Investigación en Discapacidad
70
www.medigraphic.org.mx
Introducción
La gran complejidad de los procesos biológicos en los
seres vivos ha sido por años la razón por la que los
investigadores han centrado su atención en descifrar
los mecanismos que se esconden detrás de esos pro-
cesos. Desde las primeras observaciones de Gregorio
Mendel hasta la actualidad, se tiene la noción de que
parte de la explicación de los diferentes fenómenos
biológicos se encuentra escondida en lo más recón-
dito del genoma celular y que una de las claves para
entender dichos fenómenos es el estudio de los ge-
nes. Uno de los descubrimientos más importantes de
la historia que marcó el inicio de una nueva era en el
estudio y conocimiento de los ácidos nucleicos fue el
de Watson y Crick, al descifrar la estructura del ADN
1
(ácido desoxirribonucleico). Desde entonces, varios
grupos han mostrado un gran interés por desarrollar
métodos sensibles y reproducibles que les permitan
estandarizar protocolos experimentales para estudiar
los ácidos nucleicos. Es así como han ido apareciendo
diferentes tecnologías cuyos protocolos están dirigidos
al estudio del ADN; probablemente, la más importante
sea la reacción en cadena de la polimerasa (PCR, por
sus siglas en inglés), desarrollada por Kary Mullis
1
y que
revolucionó la biología molecular y la forma en cómo
se estudiaban los ácidos nucleicos en ese momento.
Actualmente sabemos que la misión de la PCR
es copiar millones de veces una secuencia especí-
fi ca de ADN blanco mediante una poderosa catálisis
llevada a cabo por una enzima conocida como ADN
polimerasa, de tal manera que cantidades pequeñas
de ADN pueden ser sintetizadas y copiadas fi elmen-
te para analizarse con diferentes fi nes. El desarrollo
de esta técnica permitió estudiar y manipular mejor
al ADN, facilitando el establecimiento de protocolos
Fundamentos de la reacción en cadena
de la polimerasa (PCR) y de la PCR en tiempo real
Tamay de Dios L,* Ibarra C,** Velasquillo C*
* Laboratorio de Biotecnología,
Centro Nacional de Investiga-
ción y Atención a Quemados.
Instituto Nacional de Rehabi-
litación (INR), México, D.F.
** Unidad de Ingeniería de
Tejidos, Terapia Celular y
Medicina Regenerativa. INR.
Dirección para correspondencia:
M. en C. Lenin Tamay de Dios.
Avenida México-Xochimilco Núm.
289 Col. Arenal de Guadalupe,
14389, Tlalpan, México DF.
E-mail: [email protected]
Este artículo puede ser consultado
en versión completa en:
http://www.medigraphic.com/rid
Palabras clave: PCR, Taq ADN
Polimerasa, PCR en tiempo
real, SYBR Green, Taqman.
Key words: PCR, Taq DNA
Polymerase, Real Time PCR,
SYBR Green, Taqman.
Resumen
A tres décadas de su aparición, la reacción en cadena de la polimerasa (PCR, por sus siglas
en inglés) es una de las herramientas tecnológicas más innovadoras para el estudio de los
ácidos nucleicos. Se caracteriza por ser una técnica de alta sensibilidad, reproducibilidad y
eficiencia, que genera resultados confiables en poco tiempo y fáciles de analizar. Por ello,
se ha convertido en el método de elección de muchos investigadores para los estudios ge-
néticos y de biología molecular. En esta revisión nuestro objetivo es introducir al lector a los
fundamentos de la reacción en cadena de la polimerasa, así como a la reacción en cadena
de la polimerasa en tiempo real; dos técnicas que parten de un principio similar, pero que
son metodológicamente diferentes; la primera genera resultados cualitativos mientras la
segunda cuantitativos.
Abstract
After three decades of discovering utility of polymerase chain reaction, its methodol-
ogy has become one of the most employed tools for the study of nucleic acids. This is a
top sensitive, highly reproductible, very efficient and short time consuming technology
that allows to obtain reliable and easy to evaluate results. Objectives of this paper deal
with fundamentals of both techniques standard polymerase chain reaction as well as the
polymerase chain reaction on real time. Results produced by standard polymerase chain
reaction are qualitative, while those produced by polymerase chain reaction on real time
are quantitative.
Tecnología en salud
Vol. 2, Núm. 2
Mayo-Agosto 2013
pp 70-78
www.medigraphic.org.mx

Volumen 2 Número 2 Mayo-Agosto 2013
71
Tamay de Dios L y cols.
www.medigraphic.org.mx
experimentales en biología molecular. El progreso de
esta técnica ha sido muy notable y ha ido en paralelo
con los nuevos retos para estudiar y comprender mejor
el rol de los genes, tanto en condiciones fi siológicas
como patológicas. Es por ello que una de las formas
recientes para detectar y cuantifi car a los ácidos nu-
cleicos es a través de la PCR en tiempo real, la cual
es una modalidad de la PCR considerada como una
técnica cuantitativa.
Hoy en día, la PCR se aplica en diferentes áreas de
las ciencias biológicas y de la salud, formando parte
del quehacer científi co de muchos laboratorios de
investigación que la utilizan principalmente para expre-
sión génica, genotifi cación, detección de patógenos y
análisis de mutaciones. En esta revisión se hace una
descripción de los fundamentos de la PCR conven-
cional o también conocida como punto fi nal; además
se explica qué se necesita para que la reacción sea
exitosa, cómo se lleva a cabo y cómo se analizan los
resultados. Finalmente, introducimos al lector a la PCR
en tiempo real con la fi nalidad de que comprenda las
diferencias básicas con la PCR punto fi nal.
Fundamentos
¿Qué es la PCR?
La reacción en cadena de la polimerasa es una reac-
ción enzimática in vitro que amplifi ca millones de veces
una secuencia específi ca de ADN durante varios ciclos
repetidos en los que la secuencia blanco es copiada
fi elmente. Para ello, la reacción aprovecha la actividad
de la enzima ADN polimerasa que tiene la capacidad
de sintetizar naturalmente el ADN en las células. En
la reacción, si usamos como sustrato ADN genómico,
entonces típicamente hablamos de una PCR, pero si
usamos ADN complementario (ADNc) proveniente del
ARNm (ácido ribonucleico mensajero) se le conoce
como RT-PCR (Reverse Transcription-PCR, por sus
siglas en inglés). Esta conversión se logra mediante
una reacción conocida como transcripción reversa y
controlada por la enzima transcriptasa reversa, capaz
de convertir el ARNm en una molécula de ADNc. Este
método fue copiado de los retro virus que usan una
transcriptasa reversa para convertir su genoma de
ARN en ADN duplicarse en millones de partículas
virales.
2
El ADNc se utiliza cuando analizamos la ex-
presión del ARNm de algún gen de interés.
Los elementos importantes en la reacción son el
templado o molde (ADN o ADNc), la enzima, los oligo-
nucleótidos o primers, los desoxirribonucleótidos trifos-
fatados (dNTPs: adenina, timina, citosina y guanina),
el ión magnesio (Mg +), una solución amortiguadora
o buffer y H
2
O. Todos estos elementos interactúan en
tres etapas principales de las que se compone la PCR:
desnaturalización, hibridación y extensión. Los equipos
en donde se realiza la reacción son llamados termoci-
cladores, los cuales están diseñados para establecer
un sistema homogéneo en donde las condiciones de
temperatura y tiempo necesarios no se modifi quen
en cada uno de los ciclos. Al fi nal de la reacción, para
corroborar si se amplifi có la secuencia blanco de
interés, los productos de la PCR o también llamados
amplicones son analizados en geles de agarosa para
confi rmar si la reacción fue exitosa. A continuación
se explicarán cada uno de los puntos mencionados.
¿Qué elementos químicos se necesitan?
El elemento principal en la PCR es el ADN, cuya natu-
raleza química ofrece ventajas para su manipulación.
Para entrar en contexto, es importante recordar que la
molécula de ADN está formada por tres componentes:
un azúcar (desoxirribosa), un grupo fosfato y una base
nitrogenada (adenina, timina, guanina o citosina) que
es complementaria con la base de otra cadena (Figu-
ra 1), de tal forma, que el ADN se estructura en una
doble hélice.
3
La complementariedad entre las bases
está dada por puentes de hidrógeno e interacciones
hidrofóbicas, generando estabilidad a la doble hélice,
la carga eléctrica del ADN es negativa y está dada por
los grupos fosfatos. En la PCR, el templado son las
cadenas de ADN que se separan y funcionan como
molde para que la enzima sintetice las nuevas cade-
nas que llevan la secuencia blanco de interés. Por su
parte, la ADN polimerasa se encarga de la catálisis de
la reacción, sintetizando las nuevas cadenas de ADN
que llevan la secuencia blanco. La enzima más usada
con frecuencia se llama Taq ADN polimerasa,
4
que
proviene de una bacteria termófi la llamada Thermus
aquaticus, la cual vive en condiciones de temperatura
muy altas y por eso su ADN polimerasa es capaz de so-
portar ese tipo de temperaturas. El rasgo que distingue
a esta enzima bacteriana de otras ADN polimerasas
de otros organismos es su capacidad para mantener
su funcionalidad a temperaturas altas, por lo que se
le considera una enzima termoestable. También hay
otras enzimas que se utilizan como la Vent, obtenida
de la bacteria Thermococcus litoralis.
5
Normalmente,
casi todas las enzimas que se venden comercialmente
son efi cientes y generalmente cuando fallan lo hacen
por una manipulación incorrecta del usuario. Para que
la enzima funcione con alta especifi cidad y la reacción
transcurra exitosamente, también se necesita de los
Investigación en Discapacidad
72
Fundamentos de PCR y PCR en tiempo real
www.medigraphic.org.mx
elementos ya mencionados como primers, dNTPs, Mg
+, buffer y H
2
O.
Los primers son secuencias de oligonucleótidos
que fl anquean y delimitan la secuencia blanco que
se desea amplifi car y son complementarios a ésta.
Generalmente su tamaño oscila entre 15-25 pares de
bases y la cantidad de G-C no debe ser más del 55%
de la secuencia. Si no se respetan estas reglas, existe
la posibilidad de la formación de dímeros de primers,
es decir, de productos inespecífi cos. Esto repercutiría
en el rendimiento de la reacción, así como en la espe-
cifi cidad del producto esperado. Son dos secuencias
diferentes de primers las que se utilizan en la PCR,
una denominada «forward» o sentido y otra «reward»
o antisentido; ambas deben estar diseñadas para que
hibriden con el templado y las cadenas de ADN puedan
ser extendidas por la Taq polimerasa en dirección 5’-3
(como sucede endógenamente). Con la fi nalidad de
garantizar la formación de un complejo estable entre el
templado y los primers, hoy en día existen programas
informáticos para diseñar primers con alta especifi cidad,
por lo que se evita la formación de productos inespe-
rados. Incluso, hay laboratorios de biología molecular
que se dedican a diseñarlos, sintetizarlos y validarlos
para garantizar su especifi cidad, facilitándole el trabajo
al usuario que sólo elige el de su interés y los manda
a comprar. Por su parte, los dNTP’s son los ladrillos
o bases nitrogenadas con los que la Taq polimerasa
construye las nuevas cadenas de ADN. Son factores
importantes que contribuyen a la especifi cidad de la
reacción, por ello es importante que su concentración
sea la adecuada ya que de lo contrario pueden afectar la
función de la Taq polimerasa. Normalmente, se utilizan
a una concentración que oscila entre 0.2 a 1.0 mM. El
buffer es la solución amortiguadora que se usa en la
reacción y generalmente está compuesta de Tris-HCL
(pH = 8) cuya concentración fi nal de trabajo debe ser
1X. También se usan otros buffers de composición
distinta que son fácilmente comprados en el mercado.
El magnesio es un cofactor enzimático que infl uye en
la especifi cidad de la reacción, por eso se debe tener
una concentración adecuada para que no afecte el
rendimiento de la Taq polimerasa; regularmente su
concentración oscila entre 0.5 y 2.5 mM. En ocasiones
ya viene incluido en el buffer, pero en otras se le tiene
que agregar. El agua es el disolvente en la reacción y se
usa en su forma destilada libre de nucleasas, enzimas
que degradan a los ácidos nucleicos.
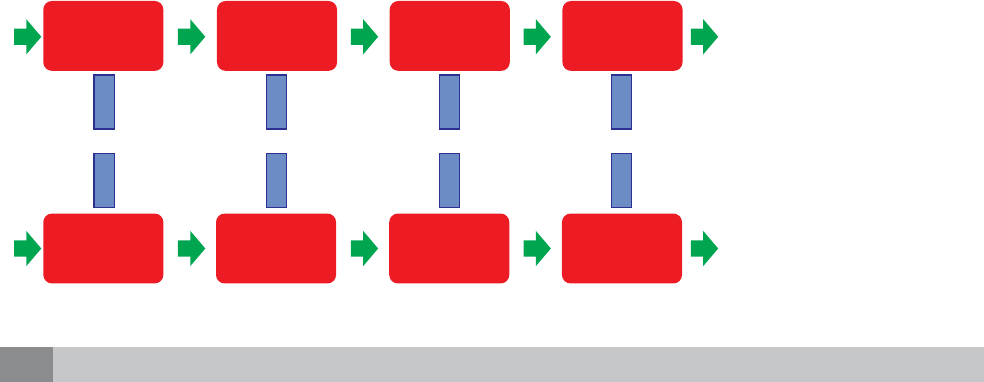
¿Cómo funciona la reacción?
Recordemos que cada ciclo de la PCR se lleva a
cabo en tres etapas principales: desnaturalización,
hibridación y extensión (Figura 2). A continuación
explicaremos qué sucede en cada una.
Desnaturalización. En esta etapa, las cadenas de
ADN son calentadas y separadas a una temperatura de
95 °C durante 20-30 segundos; el tiempo depende de
la secuencia del templado, es decir, si la cantidad de
G-C es alta, será necesario más tiempo para romper
sus uniones debido a que el apareamiento de estas
bases está formado por tres enlaces, uno más que las
bases de A-T. Además, depende de la velocidad en
la que el termociclador aumenta la temperatura, esto
varía de acuerdo al modelo del equipo. Al fi nal de esta
etapa tendremos las cadenas separadas que servirán
como templado para el siguiente paso.
Hibridación. En esta etapa, los primers se alinean
al extremo 3’ del templado previamente separado e
hibridan con su secuencia complementaria. Para que
se forme el complejo templado-primers, es importan-
te que la temperatura de hibridación o temperatura
melting (Tm) sea la óptima; ésta generalmente oscila
entre 50-60 °C. Si el diseño de los primers es el co-
Azúcar Azúcar Azúcar Azúcar
Azúcar Azúcar Azúcar Azúcar
Base
nitrogenada
Fosfato Fosfato Fosfato
ACTG
TGAC
Figura 1.
Molécula de ADN de do-
ble cadena. Cada cadena
está formada por un azúcar,
un grupo fosfato y una base
nitrogenada que es comple-
mentaria con otra cadena: A
= adenina, T = timina, C =
citosina y G = guanina.
Volumen 2 Número 2 Mayo-Agosto 2013
73
Tamay de Dios L y cols.
www.medigraphic.org.mx
rrecto y la temperatura es la adecuada, la estabilidad
y especifi cidad del complejo será efi ciente.
Extensión. En esta etapa, la Taq polimerasa actúa
sobre el complejo templado-primers y empieza su
función catalítica a una velocidad muy rápida; agre-
ga dNTP’s complementarios para crear las cadenas
completas de ADN. La extensión de las cadenas es en
dirección de la síntesis del ADN, es decir, de 5’ a 3’.
La temperatura óptima para la reacción es de 72 °C,
ya que a esa temperatura la enzima es funcional. Al
fi nal del ciclo, se habrán formado los amplicones con
un tamaño dictado por el número total de pares de ba-
ses (pb) que deberá ser conocido por el investigador.
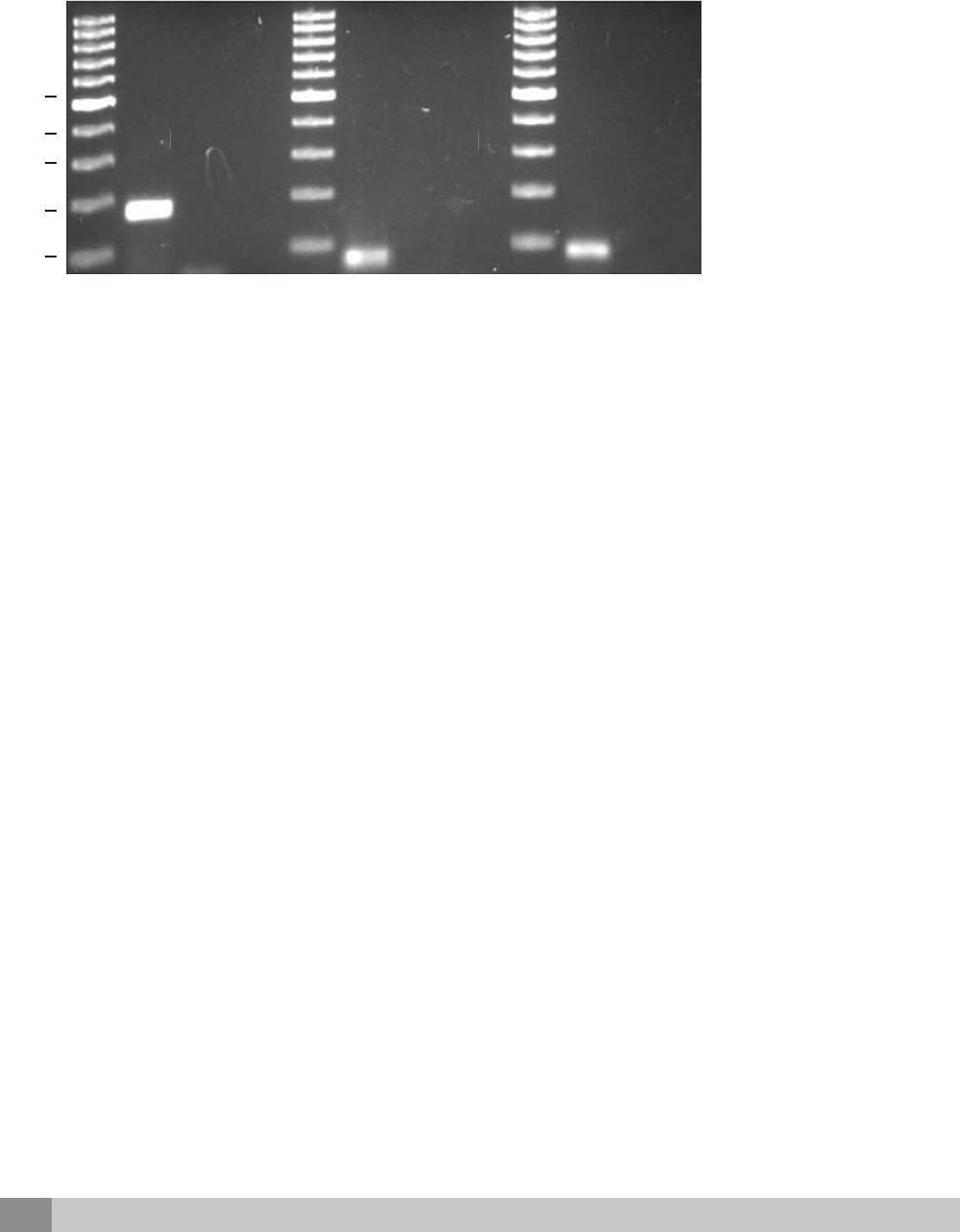
¿Cómo se analiza el producto
de amplificación?
Al fi nal de la PCR, para saber si la reacción transcurrió
efi cientemente, los amplicones son visualizados a
través de una electroforesis en geles de agarosa.
6
La
electroforesis consiste en la separación de grandes
moléculas como los ácidos nucleicos a través de una
matriz sólida que funciona como un fi ltro para separar
las moléculas en un campo eléctrico de acuerdo a su
tamaño y carga eléctrica. Esta separación se hace
bajo un buffer o tampón que puede ser TAE o TBE.
En el caso de los ácidos nucleicos, el grupo fosfato les
proporciona la carga negativa, por lo que durante la
electroforesis migran hacia el polo positivo. Para ello,
se prepara un gel diluyendo una cantidad de agarosa
en el buffer, se calienta hasta que la agarosa hierva
lo sufi ciente y posteriormente se vacía a un recipiente
que sirve de base para que solidifi que. Generalmente
el porcentaje al que se prepara el gel es al 1.2% aun-
que, dependiendo del tamaño de las moléculas, puede
ser de hasta el 2%. Otro ingrediente que se agrega
al gel es un compuesto conocido como bromuro de
etidio, una molécula intercalante capaz de unirse al
ADN de doble cadena. Cuando es excitado con luz
UV emite una señal que permite la visualización de
los amplicones en forma de bandas. Es importante
manipular con mucho cuidado este compuesto porque
se sabe que es mutagénico y teratógeno.
Cuando los amplicones son corridos en el gel, éstos
deben ser cargados junto con un marcador molecular
que contenga un número determinado de segmentos
de ADN conocidos, lo que facilita la identifi cación
de los amplicones y si su tamaño corresponde con
el esperado. El tamaño está dado por el número de
pares de pases del amplicón. El marcador de peso
molecular es fácilmente adquirido en el mercado, ya
que se ofrece una gama de marcadores con distintos
pesos moleculares para elegir el de nuestro interés.
Finalmente, la visualización de los amplicones se
lleva a cabo tomando una foto digital al gel de agaro-
sa expuesto a luz UV (Figura 3); adicionalmente un
procesador de imágenes se encarga de analizar las
bandas observadas.
La combinación adecuada de todos los elementos
químicos mencionados hacen posible la síntesis in
vitro del ADN utilizando la PCR. Los cambios que ha
sufrido la PCR como plataforma tecnológica han sido
muy notables; conforme los paradigmas de estudio de
los ácidos nucleicos han ido cambiando, la necesidad
de ir mejorando la técnica se convirtió en una prioridad
para los investigadores; es por ello que la PCR ha su-
frido modifi caciones para ofrecer una tecnología más
innovadora apegada a los retos inherentes que implica
el estudio del ADN. Actualmente, el panorama de la
PCR promete estar a la altura de los objetivos de los
investigadores, tan es así que ahora la modalidad de
la PCR en tiempo real ofrece la gran ventaja de usar
un sistema cuantitativo.
PCR en tiempo real: fundamentos
Los primeros que sentaron las bases para desarrollar
la PCR en tiempo real fueron Higuchi y colaboradores,
en 1992, al videograbar en tiempo real la incorpora-
ción de bromuro de etidio al ADN durante cada ciclo
de la PCR realizada bajo luz UV.
7
Desde entonces, el
ADN o ADNc
Primers
Taq Polimerasa
Cadena
naciente
dNTP’s
Mg+
Desnaturalización
95
o
C
Hibridación
50-60
o
C
Extensión 72
o
C
Ciclos siguientes
3’
5’
5’
3’
5’
3’
3’
5’
Figura 2. Pasos de un ciclo de la PCR.
Investigación en Discapacidad
74
Fundamentos de PCR y PCR en tiempo real
www.medigraphic.org.mx
Este documento es elaborado por Medigraphic
objetivo de la PCR en tiempo real ha sido detectar y
cuantifi car las secuencias específi cas de ácidos nu-
cleicos mediante el uso de reporteros fl uorescentes
en la reacción.
El principio de la técnica se basa en la PCR punto
fi nal, sólo que la forma en cómo se detectan y anali-
zan los productos de la amplifi cación es diferente. El
término en tiempo real se refi ere a que la detección
de los productos amplifi cados sucede en cada ciclo
de la reacción. Por su parte, el término cuantitativo
hace referencia a que es posible cuantifi car la can-
tidad de ADN en la muestra, a diferencia de la PCR
punto fi nal en donde no es posible detectar en tiempo
real ni cuantifi car la secuencia blanco. Precisamente,
estas últimas dos características representan grandes
ventajas de la PCR en tiempo real, ya que el producto
de amplifi cación es monitoreado conforme transcurre
la reacción
8
sin que haya la necesidad de que sea
manipulado en un gel de agarosa para conocer si la
reacción fue exitosa como sucede en la PCR punto
fi nal. La nomenclatura que se usa también es diferen-
te, si utilizamos ADN genómico entonces hablamos
de una qPCR (quantitative PCR), si por lo contrario,
primero obtenemos ADNc y luego hacemos la PCR,
nos referimos a una RT-qPCR.
9
Actualmente, la PCR en tiempo real es el método
más sensible para detectar y cuantifi car los ácidos
nucleicos. Aun teniendo una cantidad muy pequeña
de templado, el sistema garantiza una alta sensibilidad,
especifi cidad y efi ciencia. Una de sus aplicaciones más
usadas es para cuantifi car cambios muy pequeños en la
expresión génica mediante la detección de los niveles
del ARNm procedente de células o tejidos. La cantidad
de ARNm que puede detectar la reacción puede ser a
partir de concentraciones bajas a diferencia de la PCR,
punto fi nal que necesita una mayor concentración.
Los ingredientes químicos en la PCR en tiempo
real, son los mismos utilizados en la PCR punto fi nal,
sólo que generalmente la enzima, dNTP’s, Mg +, el
buffer y el sistema reportero de fl uorescencia para
detectar los productos amplifi cados se venden juntos
en una solución conocida como «Master mix», el
agua es proporcionada por separado y también es
libre de nucleasas. Los primers deben ser diseñados
especialmente para garantizar una alta especifi cidad
y para que generen amplicones de un tamaño que
oscile entre 100-150 pb; si éstos son más grandes, la
efi ciencia de la reacción disminuye considerablemente.
Para evitar estos problemas, una alternativa es diseñar
los primers, utilizando programas informáticos dispo-
nibles, o comprarlos ya validados de las compañías
de biología molecular, quienes garantizan resultados
altamente efi cientes y satisfactorios para los usuarios.
¿Cuáles son los métodos para detectar
los productos amplificados?
El monitoreo de los productos amplifi cados conforme
transcurre la reacción es un paso importante en la PCR
en tiempo real, para ello la estrategia tecnológica que
ha dado buenos resultados es los sistema basados en
reporteros fl uorescentes. En general, estos sistemas
pueden ser clasifi cados en dos métodos diferentes:
específi cos y no específi cos.
10
Los métodos no específi cos se basan en el uso
de moléculas intercalantes que tienen afi nidad por el
ADN de doble cadena y que al ser oxidados generan
una señal fl uorescente. La fl uorescencia emitida es
capturada en la etapa de extensión de cada ciclo y es
proporcional al número de copias de ADN de doble
cadena obtenidas en cada ciclo de la PCR. El reportero
más usado para estos fi nes se llama SYBR Green,
11
Es
Es
te
te
d
d
ococ
umum
en
en
to
e
e
s
s
el
e
el
el
l
ab
ab
ab
a
a
or
or
o
o
ad
d
o
o
po
r
r
M
e
di
d
gr
ap
h
i
c
500
400
300
200
100
PB
Marcador
conocido
Amplicón
Amplicón
Amplicón
Figura 3.
Gel de agarosa. Los produc-
tos de la PCR o amplicones
están representados median-
te bandas de un tamaño es-
pecífico y se comparan con
un marcador de peso mole-
cular conocido para deter-
minar la especificidad de la
reacción. PB = número de
pares de bases.
Volumen 2 Número 2 Mayo-Agosto 2013
75
Tamay de Dios L y cols.
www.medigraphic.org.mx
la cual es una molécula cargada positivamente que,
mientras esté en solución sin unirse al ADN de doble
cadena, prácticamente no emite fl uorescencia; sin
embargo, cuando se une al surco menor del ADN in-
crementa hasta 1,000 veces su fl uorescencia (Figura
4). Aunque el SBYR Green es uno de los reporteros
fl uorescentes más utilizados por los investigadores
debido a su bajo costo, la principal desventaja es que
puede unirse a cualquier molécula de ADN de doble
cadena, incluyendo dímeros de primers. Muchos la-
boratorios, para evitar esta situación, optimizan sus
reacciones realizando una «curva melting» o «curva
de disociación» al fi nal de la reacción, cuya función
es la de evaluar si se formó un producto único o si
hay presencia de dímeros de primers. Hoy en día la
mayoría de los software de los termocicladores ofrecen
esta función que es fácil de aplicar y analizar.
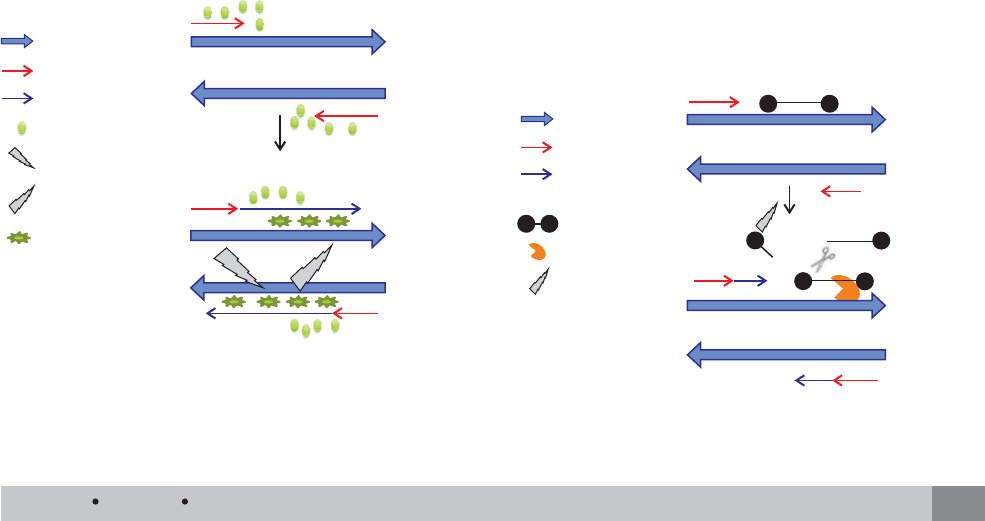
Los métodos específi cos parten de principios dis-
tintos a diferencia de los no específi cos y tienen en
común la señal de fl uorescencia emitida para detectar
los productos amplifi cados. Estos métodos siguen el
principio conocido como «transferencia de energía de
resonancia fl uorescente» (FRET, por sus siglas en
inglés) para generar la señal; este método consiste
en transferir energía desde un donador o reportero
fl uorescente a un aceptor o «quencher». Para ello,
existen dos métodos específi cos, éstos son: pruebas
basadas en hidrólisis y por hibridación. Los primeros
se basan en sondas fl uorescentes de oligonucleóti-
dos etiquetados con un reportero fl uorescente y un
«quencher», ambos se encuentran en estrecha unión
mientras la sonda no hibride a su secuencia blanco.
Cuando hibrida, ocurren cambios conformacionales en
el reportero y el quencher, lo cual permite que la acti-
vidad exonucleasa 5’-3’ de la Taq polimerasa rompa
esta unión, logrando que la fl uorescencia emitida por
el reportero sea liberada y capturada por el equipo.
Estos métodos son muy seguros, ya que mientras no
haya unión de la sonda a su blanco, no habrá am-
plifi cación y tampoco señal de fl uorescencia; es por
eso que la especifi cidad es muy alta. Un ejemplo de
estos sistemas son las sondas comerciales conocidas
como TaqMan
12
(Figura 5), aunque existen otras en
el mercado.
Los métodos por hibridación consisten en una
sonda unida a un reportero fl uorescente que está en
estrecha proximidad con un aceptor fl uorescente unido
a otra sonda. Tanto el reportero como el aceptor pre-
sentan un espectro de excitación y de emisión similar,
de tal forma que cuando las dos sondas hibriden a su
templado blanco, el reportero es excitado y la señal
emitida es transferida al aceptor, generando un incre-
mento en la cantidad de fl uorescencia. Un ejemplo de
este método son las sondas molecular Beacons
13
que
también son comerciales.
Los métodos específi cos son más costosos que los
no específi cos, pero son más efi cientes al garantizar la
especifi cidad de la reacción, evitando la formación de
productos inespecífi cos. Cualquiera que sea el método
que se aplique, se pueden adquirir fácilmente, ya que
la gama de sondas para desarrollar, tanto los métodos
específi cos como los no específi cos, es amplia.
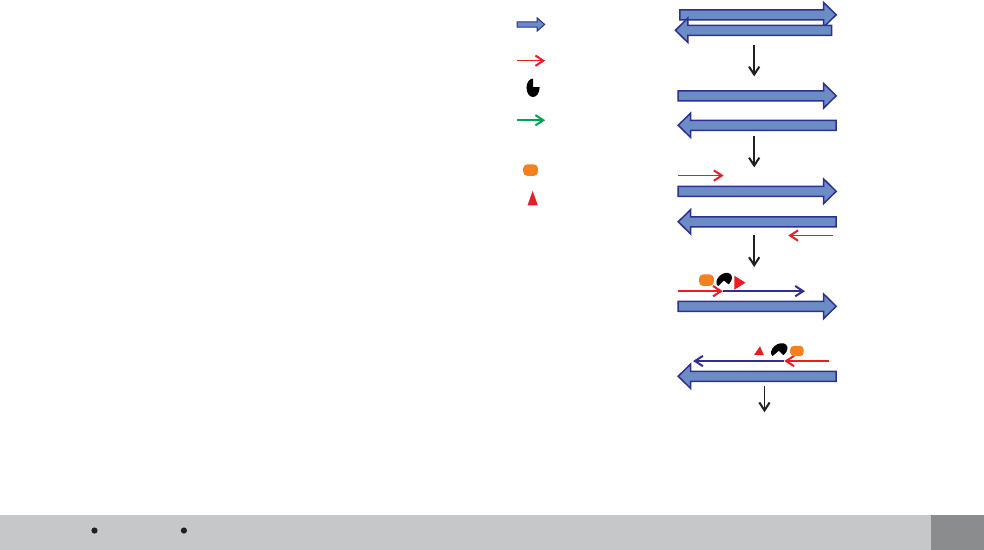
Hibridación
Extensión
ADN o ADNc
Primers
Cadena naciente
SYBR Green
Energía de excitación
Energía
de emisión
SYBR Green
fluorescente
3’
5’
3’
5’
480
520
Figura 4. Método no específico. Cuando el SYBR Green
está unido al ADN de doble cadena, es excitado a una
longitud de onda de 480 nm, mientras que la longitud
de onda de emisión corresponde a 520 nm.
Hibridación
Extensión
ADN o ADNc
Primers
Cadena
naciente
Sonda Taqman
Taq Polimerasa
Energía
de emisión
5’
3’
5’ 3’
R
Figura 5. Método específico mediante la utilización de
las sondas Taqman.
Q
Q
Q
R
R
R
Q
Investigación en Discapacidad
76
Fundamentos de PCR y PCR en tiempo real
www.medigraphic.org.mx
¿Cómo se captura la señal de fluorescencia?
Cualquiera de los métodos que se utilicen para
detectar los productos amplifi cados en cada ciclo
de la reacción necesita de la tecnología incluida en
los termocicladores de PCR en tiempo real para: a)
excitar al reportero, b) capturar la señal de emisión
del mismo y c) realizar el análisis cuantitativo. En el
mercado existen diferentes tipos de termocicladores
para esta fi nalidad, cuyas diferencias principales son
la fuente de energía que utilizan para la excitación.
En general son tres las fuentes: las lámparas de
luz, diodos de emisión de luz (LED, por sus siglas
en inglés) y láseres. Cualquiera que sea la fuente,
primero el reportero es excitado y su señal de emi-
sión colectada a través de un fi ltro que permite el
paso de la longitud de onda correspondiente que
llega hasta un fotodetector que captura la informa-
ción proveniente de la muestra para su análisis en
el software del equipo. Otros rasgos característicos
son las velocidades para incrementar o disminuir
las temperaturas en cada etapa de la reacción, el
número de muestras que puede soportar, los con-
sumibles para la reacción y los kits que se utilizan
para la amplifi cación; en algunos casos sólo se usan
reactivos del proveedor del termociclador, es decir,
son sistemas cerrados y en otros casos, se pueden
utilizar reactivos de diferentes proveedores, es decir,
son sistemas abiertos.
¿Cómo se analizan los resultados?
No es sufi ciente con detectar la amplifi cación en
tiempo real y capturar la fluorescencia de cada
muestra, el análisis de la reacción es el paso fi nal
para determinar la cuantifi cación génica. Para ello,
los termocicladores están proveídos de una PC con
un software que generalmente son fáciles de usar.
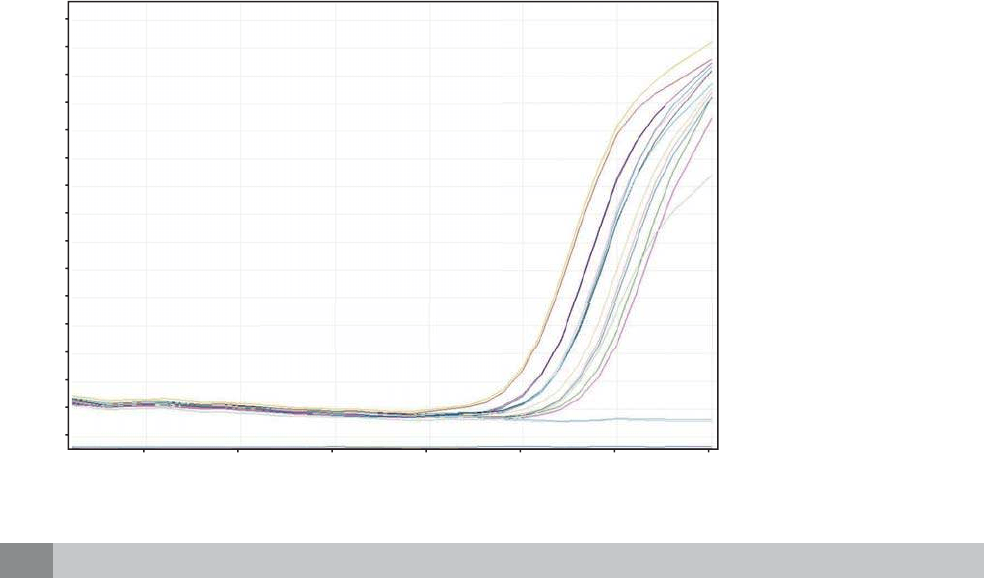
Este software genera una serie de gráfi cas en donde
se muestran todos los datos necesarios para conocer
si la reacción fue exitosa. Una de estas gráfi cas es la
de amplifi cación (Figura 6) que muestra el curso y el
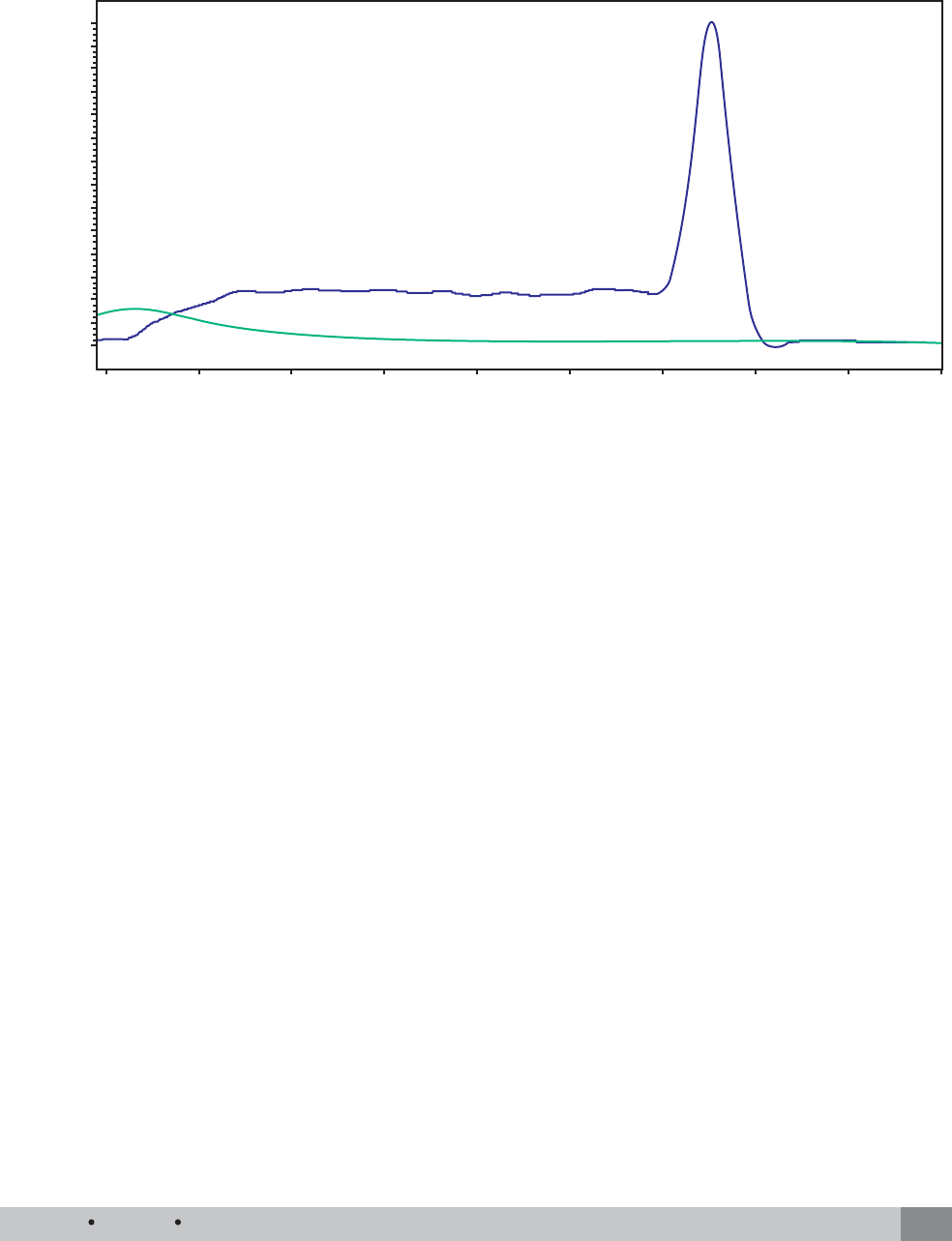
progreso de la reacción, otra gráfi ca es la curva de
disociación o curva melting (Figura 7) que muestra
información sobre la especifi cidad de la reacción.
Otro paso importante del análisis es elegir el tipo de
cuantifi cación que se usará para determinar la ampli-
fi cación precisa del blanco génico; este procedimiento
depende de los intereses del investigador. Para ello
existen dos tipos de cuantifi cación: la absoluta y la
relativa. La primera generalmente se utiliza para
conocer el número exacto de copias amplifi cadas
del blanco o la concentración precisa de ácidos
nucleicos en una muestra. En la práctica, este tipo
de cuantifi cación se usa para medir la carga viral
o bacteriana en diferentes tejidos. La segunda se
aplica cuando se desean evaluar los cambios en la
expresión de genes en distintos estados fi siológicos.
Estos cambios se basan en los niveles del ARNm del
w
w
w
w
w
.me
d
d
d
ig
ra
p
p
p
p
p
p
p
p
p
p
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
h
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
i
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
c
..
.
.
.
.
.
.
.
.
.
.
.
.
.
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
oo
o
o
o
o
o
o
o
o
o
o
o
o
o
o
o
r
r
r
r
r
r
r
r
r
r
r
r
r
r
r
r
r
r
r
r
r
r
r
r
r
r
r
r
r
r
r
r
r
r
r
r
g
.m
x
x
x
8.0
7.5
7.0
6.5
6.0
5.5
5.0
4.5
4.0
3.5
3.0
2.5
2.0
1.5
1.0
0.5
Fluorescencia
5 10 15 20 25 30 35
Ciclos
Figura 6.
Curva de amplificación. En el
eje «Y» se muestra la cantidad
de fluorescencia y en el eje
«X» los ciclos de la reacción.
La amplificación se detecta
en cada ciclo de la reacción,
midiendo el incremento de la
fluorescencia que es propor-
cional al aumento de ADN.
Volumen 2 Número 2 Mayo-Agosto 2013
77
Tamay de Dios L y cols.
www.medigraphic.org.mx
1.384
1.284
1.184
1.084
0.984
0.884
0.784
0.684
0.584
0.484
0.384
0.284
0.184
0.084
-0.016
-(d/dT) Fluorescencia (530)
50 55 60 65 70 75 80 85 90 95
Temperatura (°C)
Melting Peaks
Figura 7. Se puede observar un único pico de amplificación que corresponde a la curva de disociación o curva mel-
ting que indica la especificidad de la reacción, es decir, que los amplicones que se formaron son del tamaño espera-
do. La línea de abajo corresponde al control negativo, el cual no amplificó.
gen blanco comparados con un gen de referencia
(gen housekeeping) que no cambia su expresión a
pesar de que los estados fi siológicos se modifi quen
por diversas causas. Los datos son expresados
como relativos al gen de referencia y generalmente
son referidos como el número de veces en el que
aumentaron o decrementaron los niveles de ARNm
o en su caso, si no hubo cambios. Cualquiera que
sea el tipo de cuantifi cación que se elija, casi todos
los software de los equipos están posibilitados para
llevar a cabo los análisis matemáticos y estadísticos
que se requieren en cada tipo de cuantifi cación.
Conclusiones
Los retos que se presentan día a día en la investiga-
ción biomédica nos abren la posibilidad de conocer
nuevas herramientas tecnológicas para afrontarlos
experimentalmente. Es por eso que la PCR es una de
esas herramientas que se ha desarrollado para ofrecer
una alternativa para el estudio de los ácidos nucleicos.
Tal fue su impacto en la comunidad científi ca que hoy
en día es una técnica que se usa a diario en muchos
laboratorios y que se incrementó cuando apareció
la modalidad de PCR en tiempo real, permitiendo el
establecimiento y la aplicación de nuevos protocolos
experimentales más precisos que generan resultados
confi ables y reproducibles. Por todo ello, difundir los
fundamentos de la PCR es un primer paso para cono-
cer una de varias herramientas tecnológicas con las
que se cuenta para el estudio de los ácidos nucleicos.
Bibliografía
1. Mullis KB. The unusual origin of the polymerase chain
reaction. Sci Am. 1990; 262: 56-61.
2. Herschhorn A, Hizi A. Retroviral reverse transcriptases.
Cell Mol Life Sc. 2010; 67: 2717-2747.
3. Watson JD, Crick FH. A structure for desoxyribose
nucleic acid. Nature. 1953; 421: 397-378.
4. Saiki RK et al. Primer-directed enzymatic amplifi cation
of DNA with a thermostable DNA polymerase. Science.
1988; 239: 487-491.
5. Cariello NF, Swenberg JA, Skopek TR. Fidelity of
Thermococcus litoralis DNA polymerase (Vent) in PCR
determined by denaturing gradient gel electrophoresis.
Nucleic Acids Res. 1991; 19: 4193-4198.
6. Lee PY, Costumbrado J, Hsu CY, Kim YH. Agarose gel
electrophoresis for the separation of DNA fragments. J
Vis Exp. 2012; 62: 3923.
7. Higuchi R, Dollinger G, Walsh PS, Griffi th R. Simul-
taneous amplifi cation and detection of specifi c DNA
sequences. Biotechnology. 1992; 10: 413-417.
8. Higuchi R, Fockler C, Dollinger G, Watson R. Kinetic
PCR analysis: real-time monitoring of DNA amplifi cation
reactions. Biotechnology. 1993; 11: 1026-1030.

Investigación en Discapacidad
78
Fundamentos de PCR y PCR en tiempo real
www.medigraphic.org.mx
9. Bustin SA, Benes V, Nolan T, Pfaffl MW. Quantitative
real-time RT-PCR-a perspective. J Mol Endocrinol.
2005; 34: 597-601.
10. Foy CA, Parkes HC. Emerging homogeneous DNA-
based technologies in the clinical laboratory. Clin
Chem. 2001; 47: 990-1000.
11. Zipper H, Brunner H, Bernhagen J, Vitzthum F.
Investigations on DNA intercalation and surface
binding by SYBR green I, its structure determination
and methodological implications. Nucleic Acids Res.
2004; 32: 103.
12. Livak KJ, Flood SJ, Marmaro J, Giusti W, Deetz K. Oli-
gonucleotides with fl uorescent dyes at opposite ends
provide a quenched probe system useful for detecting
PCR product and nucleic acid hybridization. PCR
Methods Appl. 1995; 4: 357-362.
13. Tyagi S, Kramer FR. Molecular beacons: probes that fl uo-
resce upon hybridization. Nat Biotechnol. 1996; 14: 303-308.

Fundamentos de PCR y PCR en tiempo real.pdf
 Estamos procesando este archivo...
Estamos procesando este archivo...
 Lamentablemente la previsualización de este archivo no está disponible. De todas maneras puedes descargarlo y ver si te es útil.
Lamentablemente la previsualización de este archivo no está disponible. De todas maneras puedes descargarlo y ver si te es útil.
Descargar
Estamos procesando este archivo...
Lamentablemente la previsualización de este archivo no está disponible. De todas maneras puedes descargarlo y ver si te es útil.