
Examen neurológico.
Para efectuar un diagnóstico, primero debe realizarse una exploración que consta de los
siguientes puntos:
Interrogatorio o anamnesis del paciente o de sus allegados.
Examen físico: exploración de facies, actitud, marcha, motilidad, tono y trofismo
musculares, reflejos, taxia, sensibilidad, funciones cognitivas y eventualmente
estado de conciencia.
Exámenes de laboratorio: punción lumbar, con análisis del líquido cefalorraquídeo, y
otros exámenes químicos como se realizan para otros aparatos o sistemas (orina,
sangre).
Exámenes imagenológicos: tomografía computada (TC) y resonancia magnética por
imágenes (RMI), en sus diversas técnicas. Angiografía invasiva cerebral.
Exámenes electrofisiológicos: electromiografía,electroencefalografía, potenciales
evocados, potenciales motores.
Exámenes ullrasonográficos: de vasos de cuello y transcraneal, ecocardiogramas
para investigar fuente cardioembólica.
Exámenes histopatológicos: biopsias de piel, músculo, nervio y cerebro.
El objeto de la exploración del sistema nervioso es:
1. Reconocer si existe una perturbación en su funcionamiento.
2. Ver si esa perturbación depende de alteraciones anatómicas o no (orgánico vs.
funcional), y en este caso procurar localizar la región del sistema nervioso que está
alterada, o sea efectuar el diagnóstico topográfico.
3. Establecer el tipo de lesión, su mecanismo (diagnóstico fisiopatológico) y su causa inicial
(diagnóstico etiológico).
Suele comenzarse el examen del sistema nervioso por la determinación de la dexteridad,
preguntándole al paciente con qué mano escribe. En el 95% o más de las personas
diestras, que escriben con su mano derecha, la dominancia para el lenguaje es izquierda.
En cambio, aproximadamente 50% de los zurdos tienen, para el lenguaje, dominancia
derecha y 50% izquierda.
Estados de la conciencia.
Los dos extremos de la conciencia son alerta y coma, entre ambos pueden haber
alteraciones que varían en severidad o en características clínicas.
Bradipsiquia: respuestas coherentes lentificadas
Enturbiamiento de conciencia: despierto, desorientado, somnoliento.
Obnubilación: depresión de vigilia, despierta con estímulos leves
Estupor: depresión completa de estado de vigilia, despierta con estímulos intensos.

Sopor: mayor depresión del estado de vigilia responde a estímulos intensos. Mayor
“tendencia al sueño”
Coma: estado de depresión completa del estado de vigilia no despierta con ningún
estímulo.
Excitación psicomotriz: hiperactividad.
Estado vegetativo: (coma vigil-estado desaferentado) vigil con desconexion del
medio , no fija la mirada , no sigue con la mirada,ausencia de lenguaje, no responde
ordenes, reflejos de tronco conservado, ciclo vigilia sueño.Mas de 30 dias
persistente.
Mutismo Aquinetico: similar al estado vegetativo más inmovilidad absoluta, sin
signos de liberacion piramidal.
Estado de minima conciencia: deterioro de conciencia severa con minimas
respùestas a ordenes simples, gestos, movimientos, responde si - no.
Sindrome de cautiverio: no hay deterioro de la conciencia , cuadriplejia más plejia
facial bilateral solo movientos oculares.infartos bulbo-protuberanciales anteriores.
Facies.
Las enfermedades del sistema nervioso suelen mostrar cambios en los rasgos
fisionómicos.
Facies parkinsoniana: inexpresión de la cara
(cara de jugador de póker), frente fruncida por
contracción del músculo frontal, fijeza de la
mirada, falta de parpadeo, disminución o
supresión de la mímica (hipo o amimia, facies
fija o figée), seborreafacial (cara de pomada) y
boca entreabierta, con salivación pronunciada y
a veces babeo.
Facies de Hutchinson: de las oftalmoplejías
nucleares progresivas (parálisis nuclear de los
nervios bulboprotuberanciales). Se caracteriza por
párpados caídos (ptosís palpebral). El enfermo arruga
la frente y eleva las cejas para compensar la ptosís,
intentando ver a través del resquicio palpebral. Con la
progresión de la enfermedad los ojos se inmovilizan y
el enfermo no puede dirigir su mirada hacía ningún
lado.
Facies de parálisis periférica: Cuando es unilateral
presenta asimetría facial; hendidura palpebral del

lado paralizado mayor que la del lado sano, porque en el lado
enfermo la parálisis del orbicular permite una acción mayor del
elevador del párpado superior, inervado por el motor ocular
comun; el paciente no puede cerrar el ojo, y al mirar hacia
arriba su globo ocular asciende más que el del lado sano (signo
de Negro), dejando al descubierto la esclerótica (signo de Bel!);
hay lagrimeo (epifora) porque las lágrimas no se dirigen hacia
el conducto lagrimal por la parálisis del orbicular; la comisura
labial está desviada hacia el lado sano por hípotonía del lado
afectado. Cuando es bilateral la facies muestra hendidura
palpebral aumentada en ambos lados e inmovilidad de los labios, el labio inferior
pende hacía el exterior, mostrando la mucosa labial y falta la asimetría propia de la
parálisis unilateral.
Facies del síndrome de Horner: Este síndrome originado por la parálisis del plexo
simpático produce del lado afectado: ptosís parcial por parálisis del músculo liso de
Müller del párpado superior; miosis (con reflejo fotomotor preservado) por
predominio de la acción parasimpática del motor ocular común sobre el músculo
ciliar; disminución aparente de la hendidura palpebral o "hundimiento" del globo
ocular; elevación del párpado inferior (ptosis inversa) por parálisis del músculo liso
retractar de ese párpado; congestión conjuntival; falta de sudoración (anhidrosis)
frontal.
Facies de risa sardónica: Se observa en el tétanos,
hoy menos frecuente. La frente se arruga, las cejas y las
alas de la nariz se elevan, el ángulo externo del ojo se
pliega, todo lo cual da a la parte superior de la cara una
expresión de dolor; las comisuras labiales son atraídas
hacia arriba y afuera, al tiempo que los labios
contracturados descubren más o menos los dientes,
dibujando la boca en conjunto
una especie de risa permanente (risa sardónica,
espasmo cínico). Es el resultado de las contracturas
extendidas a los músculos de la cara.
Facies de miastenia gravis: Muestra signos similares a
la de las oftalmoplejías nucleares progresivas. En este
caso el trastorno se debe a bloqueo autoinmune de los
receptores postsinápticos de la placa neuromuscular. Se observa ptosis bilateral, a
veces asimétrica; el enfermo lleva su cabeza muy
hacia atrás para poder ver a través del resquicio
que deja libre la ptosis, y levanta los párpados
(mirada de astrónomo); los movimientos oculares
son lentos y se agotan fácilmente; puede haber
diplopía y estrabismo y, en casos graves,
inmovilidad ocular (ojo cuajado de Hutchinson); se asocia debilidad muscular de
labios y mejillas, con expresión característica. Lo típico en este caso es la
fatigabilidad fácil, acentuándose los síntomas a lo largo del día, o la agravación con
el esfuerzo: la ptosis se acrecienta con la elevación sostenida de los párpados.
Facies del ataque cerebral o accidente cerebrovascular: Enfermo en coma,
pléjico; rostro inmóvil, la mejilla del lado paralizado se abomba en cada movimiento
espiratorio (fumador de pipa); desviación conjugada de cabeza y ojos.
Facies del seudobulbar o suprabulbar: Hay ausencia de expresión, interrumpida
sin motivos plausibles por crisis de llanto y risa, espasmódicas e inmotivadas
(labilidad emotiva). Con frecuencia la boca está semiabierta constantemente,
dejando escapar la saliva. Se vincula a compromiso bilateral de motoneuronas
centrales ("suprabulbar").
Actitud y marcha.
La actitud de pie significa un proceso neuromuscular activo, una actividad motora estática
que requiere la cooperación de un gran número de reflejos, sobre todo tónicos. La postura
erecta se obtiene gracias a la contracción tónica de los músculos de la nuca, del tronco y
de los miembros inferiores, o sea a un predominio de los músculos del plano posterior del
cuerpo.
La marcha requiere movimientos bien coordinados, y al mismo tiempo el mantenimiento de
un equilibrio, o mejor dicho de una actitud determinada, que continuamente, a su vez,
experimentará modificaciones.
En ciertas enfermedades del sistema nervioso la actitud del enfermo de pie es muy
significativa y basta a veces para hacer el diagnóstico.
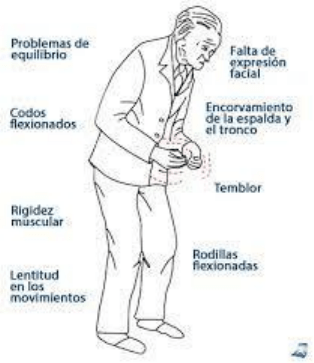
En la enfermedad de Parkinson y en los
parkinsonismos el enfermo flexiona ligeramente la
cabeza e inclina el tronco hacia adelante, con los
antebrazos en flexión y pronación y las rodillas algo
flexionadas (actitud envarada); tiene un aspecto de
fijeza, lo que unido a la ya mencionada facies
hipomímica, inexpresiva, y a la marcha descripta
más abajo, hacen inconfundibles a estos enfermos.
Tienen, además, frecuentemente temblor en las
manos y, a veces, en los brazos.
En la hemiplejía capsular con contractura, el
enfermo presenta una actitud característica:
asimetría facial (por parálisis facial inferior), el miembro superior paralizado,
apoyado todo contra el tórax, con el antebrazo flexionado sobre el brazo y en
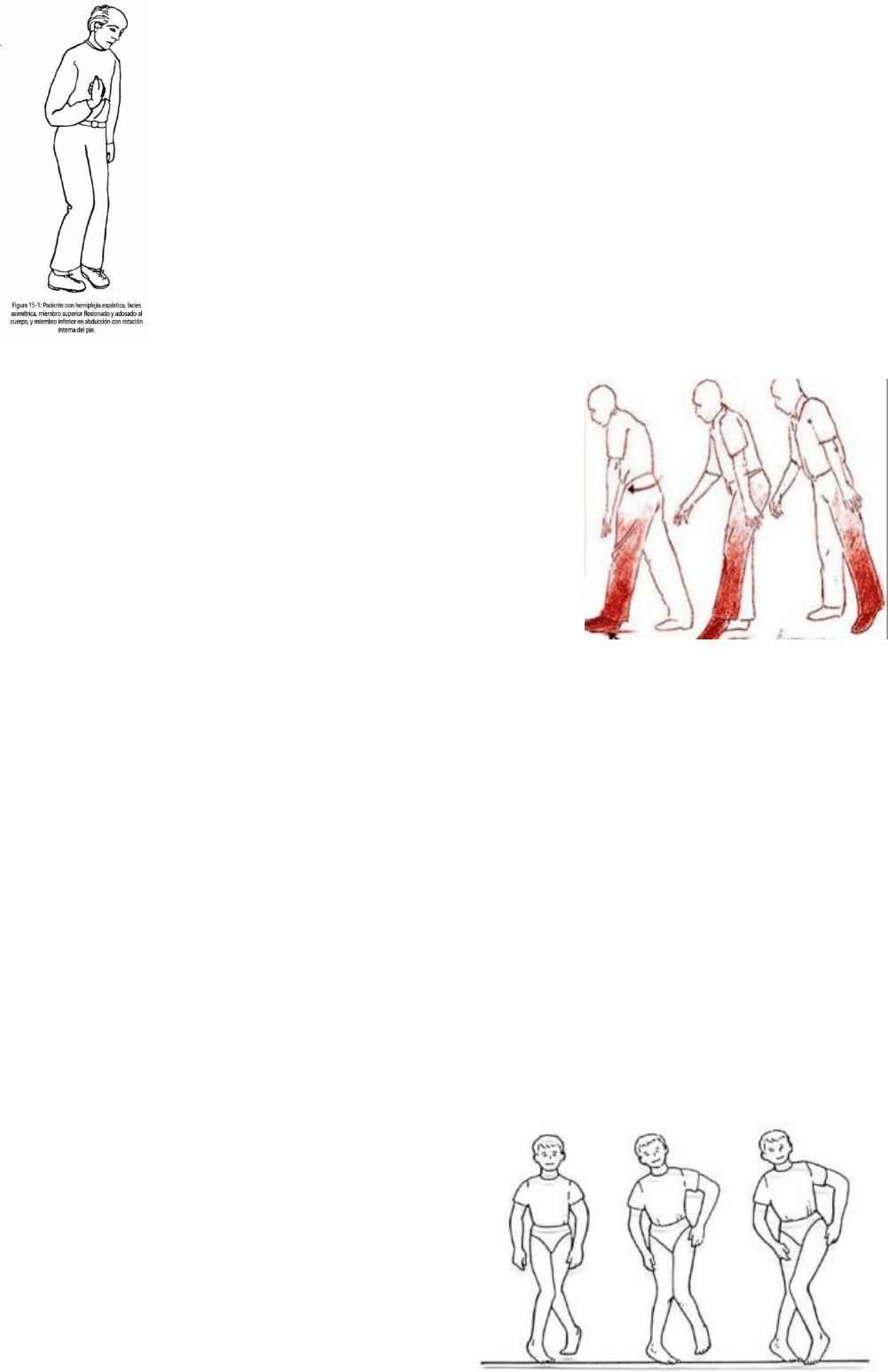
pronación, la mano flexionada sobre el antebrazo, los dedos de la mano
sobre la palma y el pulgar con frecuencia aprisionado bajo los otros
dedos. El miembro inferior paralizado se halla recto, con cierto grado de
aducción y, a veces, de inversión o rotación interna del pie.
Para evaluar la marcha, se le pide al paciente que camine descalzo en
línea recta.
Marcha paretica: Se debe a la paresia de los músculos de
los miembros inferiores. A consecuencia de la parálisis de los extensores
del pie y peroneos, el pie cuelga y su punta roza el suelo al andar. El
enfermo se ve obligado, para no tropezar, a
levantar ampliamente la pierna a cada paso,
doblando el muslo sobre la pelvis y elevando
los pies sin extenderlos o flexionarlos
dorsalmente. Luego los pies caen al suelo
apoyando primero la punta de los dedos y
después el borde externo. Se observa en
afecciones del asta anterior medular como la
esclerosis lateral amiotrofica o secuela de
poliomelitis aguda.
Marcha pareticoespástica: se produce aumento del tono muscular
acompañando a la paresia. Se debe a lesión de la vía piramidal, y predomina
el carácter espástico. Se observa:
En hemiplejia capsular, avanza trazando con el miembro inferior enfermo un semicírculo,
arrastrando el pie que apoya sobre el suelo por su borde externo y punta (marcha
helicoidal). La punta del pie se desprende del suelo con dificultad debido a la hipertonía, y
el calzado se gasta mucho en la punta.
En las paraplejías espásticas de diverso origen (esclerosis múltiple, compresiones
medulares, familiares), el enfermo camina con los miembros inferiores estirados y apenas
puede levantar la punta de los pies. Da pasos pequeños, rozando el suelo con la parte
anterior o anterointerna de los pies, gastando la punta de sus zapatos.
En las parálisis cerebrales congénitas, el enfermo camina cruzando una pierna delante de
la otra en X, con pasos cortos. Es la marcha en tijera. Se debe a hipertonía de los
aductores y desgasta la ropa en las rodillas,
por frotamiento.
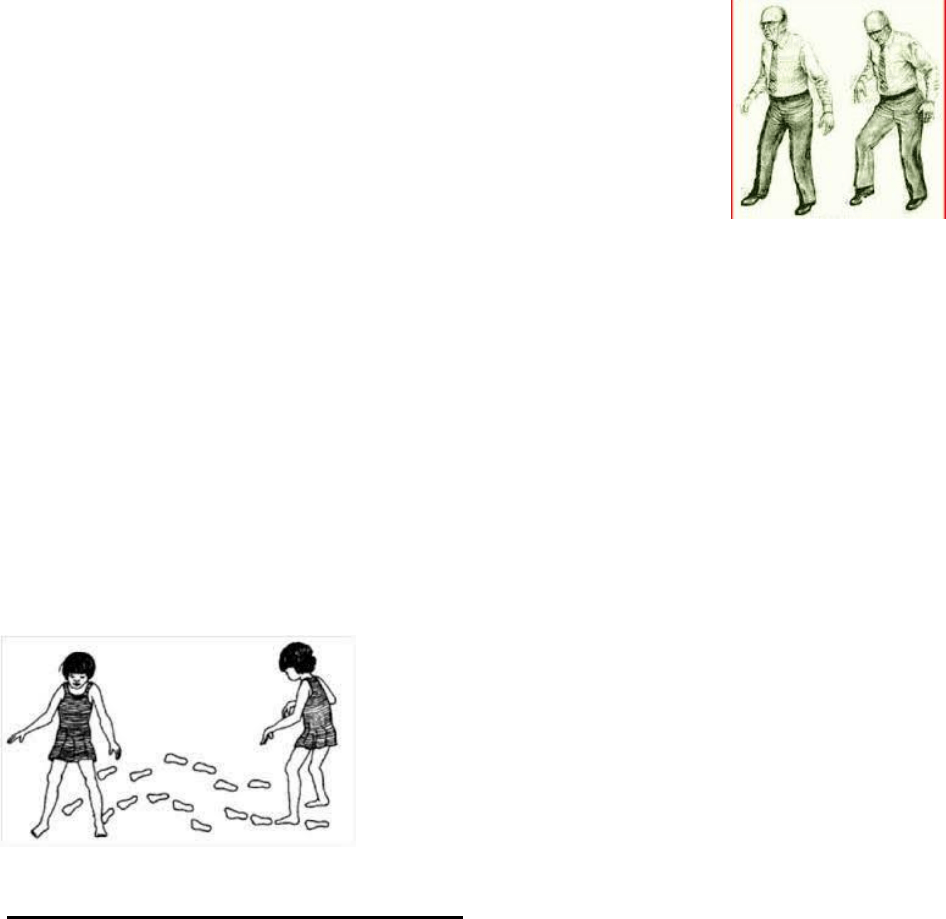
Marcha ataxica: Se debe a trastorno de la taxía (coordinación motora), sin paresia
ni hipertonía. Hay aumento de la base de sustentación (piernas separadas) e
inestabilidad, vacilación y falta de medida en los movimientos. Aparece en caso de
lesiones del aparato regulador de la coordinación motora:
nervios (neuropatlas periféricas o polineuropatías),
cordones posteriores de la médula (enfermedad de
Friedreich y, clásicamente, tabes dorsal por neurosífilis),
cerebelo (atrofias y tumores del cerebelo) y laberinto
(laberinlitis). Pueden sutilmente distinguirse tres tipos: la
marcha "tabética", la cerebelosa y la tabetocerebelosa,
combinación de ambas.
En la marcha tabetica, el paciente marcha separando sus piernas con exceso, mirando al
suelo, levantándolas súbita y violentamente para proyectarlas con energía sobre el suelo,
cayendo sobre el talón (taloneo). La mirada hacia abajo tiene por objeto corregir la ataxia
con la vista y apreciar mejor las distancias. Las lesiones de los cordones posteriores
impiden el arribo de impulsos propioceptivos (de músculos, articulaciones, ligamentos y
huesos) a los centros coordinadores del cerebelo.
En la marcha cerebelosa, hay titubeo en el andar. En la estación de pie puede observarse
a los músculos extensores de los pies, inestables, contraerse continuamente y a destiempo
(baile de los extensores). El enfermo camina vacilando, con tendencia a desplazarse o
caer hacia los lados (lateropulsión), hacia adelante (propulsión), o hacia atrás
(retropulsión). Al ordenársele avanzar en línea recta, se
desvía de su trayectoria, en zigzag.
La marcha tabetocereblosa es una marcha tabetica con
zigzag.
La marcha atáxica se combina a veces con la espástica:
es la marcha ataxicoespástica, en la que el enfermo
titubea y arrastra los pies.
Evaluación de los pares craneales.
I par olfatorio.
Origen real: células olfatorias de la mucosa olfatoria.
Origen aparente: cara inferior del bulbo raquídeo.
Se explora presentando un olor común, fácil de reconocer (café, oregano, vainilla). Se
acerca a una narina y se ocluye la contraria, y luego se invierte.
Las alteraciones son:
-Hiposmia
-Anosmia
-Hipersosmia.
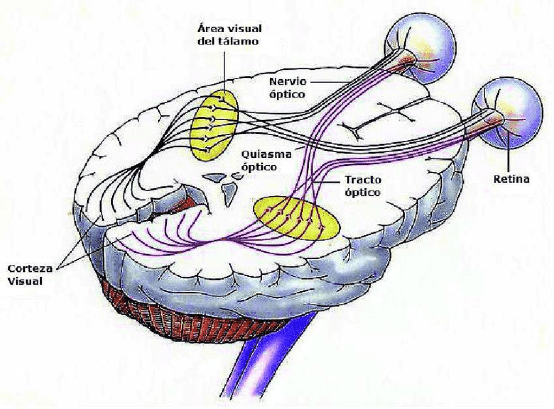
II par óptico.
Origen real: células ganglionares de la retina.
Origen aparente: ángulo anteroexterno del quiasma óptico.
Se evalúa agudeza visual, a distancia y de cerca, los campos visuales, el fondo de ojo y
la visión de colores. También se evalúa la pupila con estimulación de la luz.
Cuando hay lesión del nervio óptico se da escotoma (mancha negra que deja la
inflamación del óptico porque se aumenta el punto ciego), se pierde la visión central o toda
la visión dependiendo de donde se dé el daño.
Las señales nerviosas viajan desde cada ojo a través del nervio óptico correspondiente y
de otras fibras nerviosas (denominadas vías ópticas) hasta la parte posterior del cerebro,
donde se percibe y se interpreta la visión. Los dos nervios ópticos se unen en el quiasma
óptico, una zona situada detrás de los ojos, justo delante de la hipófisis y debajo de la
porción frontal del encéfalo (cerebro). En este lugar, el nervio óptico de cada ojo se divide y
la mitad de las fibras nerviosas de cada lado cruzan al otro lado y continúan hasta la parte
posterior del cerebro. Así, el lado derecho del cerebro recibe información, a través de
ambos nervios ópticos, del campo visual izquierdo, y el lado izquierdo del cerebro recibe
información, a través de ambos nervios ópticos, del campo visual derecho. La parte central
de dichos campos de visión se superpone, y la ven los dos ojos (lo que se conoce como
visión binocular).
III, IV y VI par nervios motor ocular común, patético y motor ocular externo.
Comparten la inervación de los músculos extrínsecos del ojo, por lo que corresponde
estudiarlos en conjunto. Todos ellos están constituidos por fibras eferentes motoras.
Origen real III: núcleo somatomotor y núcleo parasimpático motor.
Origen aparente III: cara interna de los pedúnculos cerebrales.
Origen real IV: núcleo somatomotor.
Origen aparente IV: cara posterior de los pedúnculos cerebrales.
Origen real VI: núcleo protuberancial por debajo del piso del IV ventrículo.
Origen aparente VI: surco bulboprotuberancial.
Se evalúa tamaño y forma de la pupila, relfejo fotomotor y consensual, acomodación
pupilar y convergencia.
También se debe evaluar ptosis palpebral y movimientos oculares.
Realiza una H con los ojos.
V trigémino.
Tiene su origen aparente en la región anterior y lateral de la protuberancia, cerca del
pedúnculo cerebeloso medio y sus orígenes reales distribuidos en las neuronas
pseudounipolares.
Se evalúa el reflejo corneano, la sensación del rostro con algodon, la fuerza del musculo
masetero.
VI facial.
El origen real de las fibras motoras está en el núcleo motor del nervio facial; el de las fibras
sensitivas en el ganglio geniculado. Su origen aparente se encuentra en el tronco
encefálico, específicamente en el surco bubloprotuberancial.
Se evalúa elevación de las cejas, si arruga la frente, si puede cerrar los ojos, sonrisa, que
infle los cachetes.
Se debe evaluar el gusto en los 2/3 anteriores de la lengua, con sabores dulces, salados,
amargos.
VIII par auditivo.
Las raíces emergen del origen real del nervio, que son los núcleos vestibulares y
cocleares ubicados en el tronco encefálico. Estos salen del mismo por el surco
bulbopontino ubicado entre el puente y el bulbo raquídeo, sitio que representa al origen
aparente del nervio.
Se evalúa con el chaqueo de los dedos o frotar el pelo del paciente.
IX glosofaríngeo y X vago.
Origen aparente IX: surco lateral del bulbo.
Origen real IX: Fibras sensitivas: nacen del ángulo externo de la sustancia reticulada
gris. Fibras motoras: nacen del núcleo ambiguo
X: Su origen real se encuentra en las células del ganglio petroso, que terminan a nivel
del tracto solitario del bulbo raquídeo. Su origen aparente está entre los nervios
craneales accesorio (XI) y glosofaríngeo (IX), en el surco colateral posterior del bulbo
raquideo o surco retroolivar.
Se evalúa la simetría del paladar y la úvula diciendo “aaa”, la sensibilidad posterior de
la faringe y la lengua y el reflejo nauseoso.
XI espinal.
Origen real: Núcleo bulbar: ubicado en las células de la porción inferior del núcleo
ambiguo.
Origen aparente: surco colateral posterior del bulbo raquídeo.
Se evalúa elevando los hombros y rotando la cabeza contra resistencia.
XII Hipogloso.
Tiene su origen real eferente somático general en el núcleo motor, localizado en el bulbo
raquídeo, y su origen aparente en el surco preolivar.
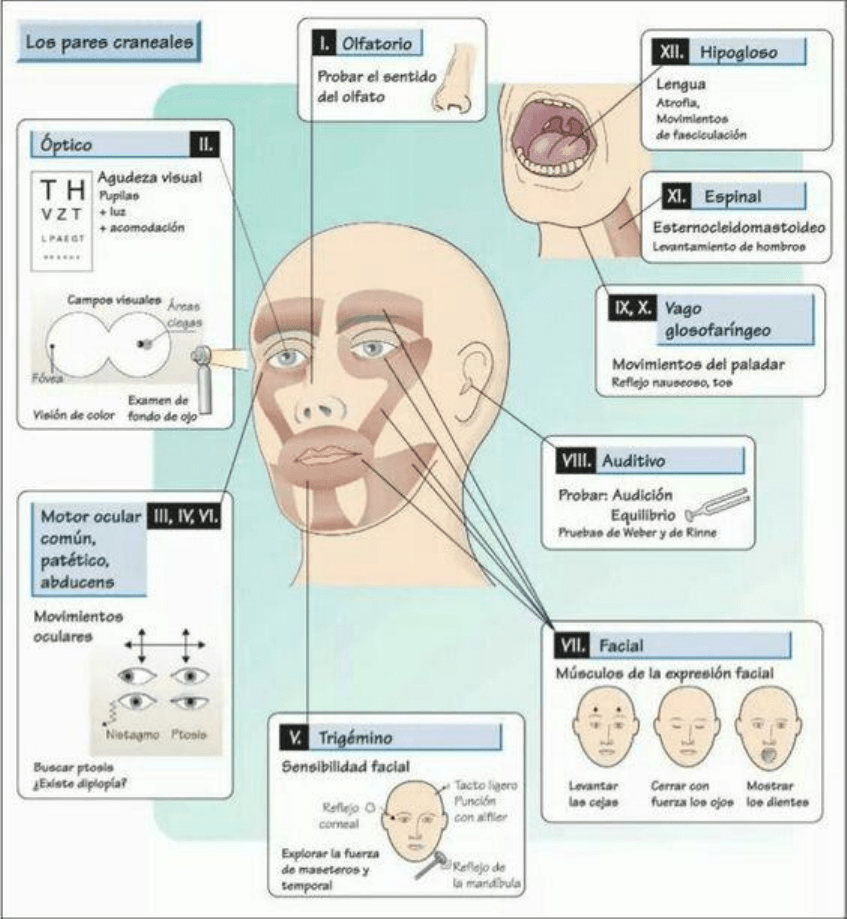
Se evalúa la lengua buscando anomalías, su fuerza y atrofias.
Tono muscular.
Un músculo vivo, correctamente inervado y vascularizado y en ausencia de motilidad
voluntaria, se halla en un ligero grado de contracción que constituye un estado de
semítensión particular, que no es ni la flaccidez de un músculo separado de sus
conexiones, ni la tensión fuerte y dura de un músculo en contracción activa. Ese estado
particular del músculo en reposo es el tono muscular.
El tono es esencialmente la actividad postural de la musculatura. En su actividad tónica los
músculos funcionan, no como una máquina que produce trabajo mecánico, sino como
aparatos fijadores de los segmentos óseos y cartilaginosos del esqueleto.
Los estímulos propioceptivos que aseguran el tono muscular son recogidos por los husos
neuromusculares, corpúsculos fusiformes situados entre las fibras musculares. Constituyen
los órganos receptores sensibles al estiramiento, estando constituidos por 4 a 12 fibras
fusales de diferente diámetro que poseen inervación propia a través de las motoneuronas y
(gamma), incorporadas al sistema de la motoneurona periférica, cuya célula de origen está
en el asta anterior medular. La mayor o menor descarga de estas motoneuronas y produce
una mayor o menor tensión de las fibras del huso neuromuscular. A mayor actividad y,
mayor tensión del huso, y viceversa. A mayor tensión del huso, mayor resistencia al
estiramiento, y viceversa.
Los estímulos generados por el estiramiento de los husos siguen por fibras sensitivas
gruesas llegando a la médula para hacer sinapsis en las motoneuronas alfa que, al
estimularse, determinan la contracción de las fibras musculares. Esta actividad constituye
un arco reflejo: el reflejo miotático.
Paralelo al sistema del huso neuromuscular existe otro sistema, el de Golgi, constituido por
los órganos de este nombre, igualmente sensibles al estiramiento. Se localizan en los
tendones, a nivel de su unión con los músculos, que, al ser estimulados, descargan sobre
la motoneurona alfa, su umbral es de excitación mayor que el del huso. Por lo tanto, este
sistema es menos sensible que el de los husos, y actúa inhibiendo el reflejo miotático.
El paleocerebelo, por intermedio del núcleo rojo y el haz rubroespinal cruzado, ejerce una
función inhibidora sobre los centros espinales del tono.
El neocerebelo obra sobre el tono muscular en sentido dinamógeno o facilitador, pero no
funciona sino en asociación con la corteza cerebral.
Finalmente, la corteza cerebral tiene una acción evidente. Se admite corrientemente que el
área motriz cortical y la vía piramidal ejercen una acción inhibidora sobre el tono muscular.
Por ello una lesión piramidal determina hipertonía.
Si se lesionan las vías aferentes sensitivas o sensoriales, o si se destruyen los centros
tonígenos medulares o supramedulares, así como la vía motriz periférica, se producirá
hipotonia. Por ello existe hipotonía en la polineuritis, en las lesiones del cordón posterior
medular, en la poliomielitis y en las afecciones cerebelosas.
Si en cambio la lesión afecta a los centros o vías inhibidoras del tono, el resultado será que
los centros subyacentes liberados exageren su acción y se produzca hipertonía. Tal es el
caso de las lesiones de la vía piramidal y del locus niger.
Taxia.
Todos los músculos actúan normalmente como una unidad. Su actividad se gradúa y
ajusta recíprocamente: si el agonista exagera su contracción, el antagonista se relaja, y
los sinergistas y fijadores acentúan su acción. El resultado es un movimiento adecuado
al fin propuesto: un movimiento coordinado
La necesidad de coordinación ha sido demostrada por la neuropatología: muchos
enfermos tienen trastornos motores sin parálisis o paresia, pero con perturbación de la
coordinación, llamada ataxia.
Primero, intervienen las vías sensitivas aferentes que conducen al cerebro impulsos
propíoceptivos originados periféricamente en receptores cinestésicos por
desplazamiento y distensión mecánica de músculos, tendones, articulaciones y partes
blandas. Estos impulsos se conducen a la médula espinal por fibras aferentes
sensitivas de los nervios periféricos y de la raíz posterior medular, originadas en el
ganglio anexo a dicha raiz (primera neurona de la vía). Llegadas a la médula las fibras
siguen diversos caminos.
1. Vía cordonal posterior:
Propioceptiva consciente.
Cruzada, 3 neuronas.
Aferencia periférica (axón 1): receptor, nervio periférico,
raíz dorsal.
Neurona I: ganglio de la raíz dorsal.
Axón II: prolongación proximal de la neurona 1, fascículos gracilis (niveles sacros a D6),
cuneiforme (D5 a cervicales).
Neurona II: núcleos gracilis y cuneiforme.
Decusación
Axón III: lemnisco medial.
Neurona III: núcleo ventral posterolateral del tálamo.
Axón IV: proyección talamoparietal.
Punto final: corteza parietal.
2. Vía espinocerebelosa ventral:
Propioceptiva inconsciente.
Cruzada, 2 neuronas.
Aferencia periférica (axón 1): receptor, nervio periférico, raíz dorsal.
Niveles de proveniencia: miembros inferiores y mitad inferior del tronco.
Neurona I: ganglio de la raíz dorsal.
Axón II: prolongación proximal de la neurona l.
Neurona II: asta posterior.
Decusación
Axón III: fascículo espinocerebeloso ventral.
Pedúnculo cerebeloso superior.
Punto final: paleocerebelo.
3. Vía espinocerebelosa dorsal:
Propioceptiva inconsciente.
Directa, 2 neuronas.
Aferencia periférica (axón I): receptor, nervio periférico, raíz dorsal.
Niveles de proveniencia: tronco y pelvis (T1-L2).
Neurona I: ganglio de la raíz dorsal.
Axón II: prolongación proximal de la neurona l.
Neurona II: núcleo dorsal de Clarke.
Axón III: fascículo espinocerebeloso dorsal.
Pedúnculo cerebeloso inferior.
Punto final: paleocerebelo.
4. Vía cuneocerebelosa:
Propioceptiva inconsciente.
Directa, 2 neuronas.
Aferencia periférica (axón I): receptor, nervio periférico, raíz dorsal.
Niveles de proveniencia: miembros superiores y cuello.
Neurona I: ganglio de la raíz dorsal.
Axón II: prolongación proximal de la neurona l.
Neurona II: núcleo cuneiforme lateral.
Axón III fascículo cuneocerebeloso (fibras arciformes externas dorsales).
Pedúnculo cerebeloso inferior.
Punto final: paleocerebelo.
Ataxias periféricas
Se observa en las neuropalías periféricas o polineuropatías. La ataxia radicular es de
observación sumamente raras. La ataxia periférica se debe a la imposibilidad de los
estímulos propioceptivos de alcanzar el sistema nervioso central. Se la denomina también
ataxia neurítica.
En los miembros hay trastornos tróficos distales, hiporreflexia o arreflexia profunda,
hipoestesia "en bota" o "en guante". En raros casos la sensibilidad superficial está intacta
existiendo sólo alteraciones de la sensibilidad profunda (abatiestesia: hipoestesia para las
actitudes segmentarías). Las masas musculares y troncos de nervios periféricos pueden
doler con la presión. No suele acompañarse de signo de Romberg. Cuando ello sucede, se
la denomina seudotabes periférica porque el cuadro se asemeja al clásico de la tabes
dorsal de origen luético.
Ataxias medulares.
Se manifiesta en las mielopatías que lesionan los cordones posteriores. Se produce ataxia,
que se origina por la falta de conducción de las impresiones propioceptivas periféricas a los
núcleos encefálicos de la coordinación motriz, y trastornos sensitivos (hipoestesia o
anestesia profunda y táctil epicritica o discriminatoria). La ataxia es estática (signo de
Romberg) y dinámica.
Se observa en la esclerosis múltiple, en la enfermedad de Friedreich y otras
degeneraciones espinocerebelosas, en la degeneración combinada, en la siringomielia, la
mielopatía cervical por canal estrecho y, clásicamente, en la tabes dorsal.
Ataxia cerebelosa.
La ataxia cerebelosa resulta de varios factores: 1 º pérdida de la eumetría: el movimiento
efectuado carece de medida (dismetría); 2 º falta de sinergia (asinergia) y 3 º falta de
coordinación en el tiempo (adiadococinesia), de los distintos músculos al realizar el
movimiento. El cerebelo controla todos estos factores, que se alteran cuando el mismo se
lesiona. En la estación de pie el cerebeloso oscila en todo sentido, lentamente, aunque no
suele caer (ataxia estática). El cierre de los ojos no agrava su desequilibrio (Romberg
ausente). La marcha es disbásica, con piernas separadas para aumentar la base de
sustentación (ataxia dinámica). La asociación de reflejos pendulares, temblor intencional,
nistagmo, pruebas de pasividad, disartria, todos signos de la serie cerebelosa, permite
diferenciar la ataxia cerebelosa de los otros tipos de ataxia.
La ataxia cerebelosa se ve en las lesiones cerebelosas o de sus vías.
Las vías aferentes al cerebelo se dividen en resumen en cinco grupos principales, según
su punto de partida: a) espinales (núcleo de Clarke y asta posterior), b) vestibulares
(núcleos vestibulares), c) reticulares (núcleos bulbopontinos), d) olivares (oliva bulbar) y e)
pontinas (núcleos del puente). Pero, aparte de estos cinco grupos principales de
aferencias, hay otros numerosos fascículos de fibras que alcanzan el cerebelo a partir de
diversas estructuras del tronco cerebral, que reciben aferencias sensoriales visuales y
auditivas.
Puede afirmarse que el cerebelo ejerce sobre la motilidad cinética y estática una aclívidad
reguladora que se sintetiza así:
- Asegura la eumetría e isostenia de los movimientos, o sea, hace que el movimiento tenga
la necesaria intensidad o fuerza y la exacta medida que requiere el fin buscado.
-Asegura la sinergia y la diadococinesia (, es decir la coordinación de los diversos grupos
musculares, cuando deben contraerse para realizar esos movimientos.
- Regula el tono muscular.
- Interviene en el mantenimiento de la postura y del equilibrio.
Síndrome cerebeloso.
A veces aparece vértigo cuando el enfermo se encuentra en bipedestación, en decúbito
horizontal o lateral y en este caso suele producirse cuando el enfermo está acostado sobre
el lado opuesto al de la lesión.
Las manifestaciones pueden ser estáticas, cinéticas, de los movimientos pasivos y otras.
Ataxia laberíntica.
En la estación de pie, el paciente lateropulsa hacia el lado afectado. Al ocluir los ojos, la
lateropulsión se acentúa, eventualmente hasta la caída (signo de Romberg laberíntico). Si
se hace rotar la cabeza, la lateropulsión se produce en el eje anteroposterior. En la marcha
no hay verdadera ataxia, pero el laberíntico la desvía hacia el lado afectado cuando camina
hacia adelante, y hacia el lado opuesto cuando lo hace hacia atrás.
Ataxias episódicas.
Son aquellas que aparecen de manera súbita, y pueden deberse a causa tóxica, esclerosis
múltiple y la forma basilar de la migraña (Bickerstaff). Integran también este grupo las
ataxias episódicas de herencia autosómica dominante.
Este documento contiene más páginas...
Descargar Completo
Examen neurológico.pdf
 Estamos procesando este archivo...
Estamos procesando este archivo...
 Lamentablemente la previsualización de este archivo no está disponible. De todas maneras puedes descargarlo y ver si te es útil.
Lamentablemente la previsualización de este archivo no está disponible. De todas maneras puedes descargarlo y ver si te es útil.
Descargar
Estamos procesando este archivo...
Lamentablemente la previsualización de este archivo no está disponible. De todas maneras puedes descargarlo y ver si te es útil.