
© Elsevier. Es una publicación MASSON. Fotocopiar sin autorización es un delito.
7
115
Capítulo
Patrones de herencia monogénica
En el capítulo 1 se expusieron brevemente las tres categorías
principales de trastornos genéticos, los monogénicos, los cro-
mosómicos y los complejos. En este capítulo se van a explicar
con detalle los patrones característicos de transmisión de los
trastornos monogénicos, con insistencia en los mecanismos
moleculares y genéticos a través de los cuales las mutaciones
en los genes dan lugar a los patrones de herencia recesiva, do-
minante, ligado al cromosoma X y mitocondrial. En el capítu-
lo siguiente se describirán los patrones más complejos de he-
rencia, incluyendo los trastornos multifactoriales que se deben
a interacciones entre las variantes en loci múltiples y a factores
ambientales que causan enfermedad.
Los rasgos monogénicos causados por las mutaciones en
los genes del genoma nuclear se denominan a menudo men-
delianos debido a que, de la misma forma que los típicos gui-
santes cultivados que fueron estudiados por Gregor Mendel,
aparecen en proporciones fi jas entre los descendientes de tipos
específi cos de emparejamientos. Las enfermedades monogéni-
cas conocidas hasta el momento están recogidas en la referen-
cia clásica de Victor A. McKusick, Mendelian Inheritance in
Man, que durante decenios ha constituido un elemento indis-
pensable para los especialistas en genética médica. La versión
en línea de Mendelian Inheritance in Man (OMIM, online ver-
sion of Mendelian Inheritance in Man), a la que se puede ac-
ceder en Internet a través de la National Library of Medicine,
recoge en la actualidad más de 3.917 enfermedades con patro-
nes de herencia mendeliana. De ellas, 3.310 (aproximadamen-
te, el 84%) se deben a mutaciones en 1.990 genes. El número
de enfermedades con causas genéticas conocidas y el núme-
ro de genes cuyas mutaciones pueden causar enfermedad no
son iguales debido a que las mutaciones diferentes en un mis-
mo gen pueden dar lugar a enfermedades distintas, y a que las
mutaciones en diferentes genes pueden causar enfermedades
similares o indistinguibles entre sí. El 16% de las enfermeda-
des recogidas en la OMIM corresponde a trastornos con pa-
trones de herencia claramente mendeliana, pero respecto a los
cuales todavía se desconocen los genes responsables. Así, de
los aproximadamente 25.000 genes humanos, alrededor del
8% ya ha sido implicado directamente en enfermedades gené-
ticas del ser humano. Es posible que ésta sea una estimación
groseramente insufi ciente. El ritmo con el que los especialistas
en genética están identifi cando genes con alelos causantes de
enfermedad es muy elevado y –ciertamente– se ha acelerado a
consecuencia de las nuevas y potentes herramientas derivadas
del Proyecto Genoma Humano.
En conjunto, los trastornos monogénicos se observan prin-
cipalmente en el rango de edad pediátrico, aunque de ninguna
manera son cuadros patológicos exclusivos de esta banda de
edad; menos del 10% se manifi esta después de la pubertad y
tan sólo el 1% lo hace hacia el fi nal del periodo reproductivo.
A pesar de que individualmente son raros, en conjunto estos
trastornos son los responsables de una proporción signifi cativa
de enfermedades y fallecimientos infantiles. En un estudio de
población efectuado sobre más de 1 millón de recién nacidos
vivos, se estimó que la incidencia de trastornos monogénicos
graves era del 0,36%; entre los niños hospitalizados, se conside-
ró que el 6-8% sufría algún trastorno monogénico. También es
importante considerar los trastornos mendelianos en la medici-
na del adulto. En un estudio efectuado sobre la OMIM respecto
a las formas mendelianas de 17 de las enfermedades del adulto
más frecuentes (como la cardiopatía, el accidente cerebrovas-
cular, el cáncer y la diabetes), se detectaron casi 200 trastornos
mendelianos en cuyos fenotipos se incluían estas enfermedades
frecuentes del adulto. A pesar de que las formas mendelianas no
son de ninguna manera el factor contribuyente principal para
la aparición de estas enfermedades comunes en la población ge-
neral, tienen importancia en los pacientes individuales debido a
su signifi cación para la salud de los familiares de los pacientes y
debido también a la existencia de pruebas genéticas y de opcio-
nes terapéuticas concretas en muchas de ellas.
PANORÁMICA GENERAL Y CONCEPTOS
A pesar de que los principios de la genética médica son de fácil
compresión, la difi cultad con la terminología puede hacer que
inicialmente estos principios parezcan inaccesibles. Para facili-
tar la solución del problema con el lenguaje, a continuación se
van a revisar algunos términos y se van a introducir otros que
no han sido defi nidos previamente.

Thompson & Thompson GENÉTICA EN MEDICINA
116
Variación en los genes
La variación hereditaria en el genoma representa la piedra an-
gular de la genética humana y médica. Tal como se ha descrito
en el capítulo 2, un segmento de DNA que ocupa una posi-
ción o localización concretas en un cromosoma es un locus.
Si este segmento contiene un gen, dicho segmento de DNA es
el locus para este gen. Las variantes alternativas de un gen se
denominan alelos. En lo que se refi ere a muchos genes, hay un
único alelo predominante que aparece en la mayor parte de los
individuos y que los especialistas en genética denominan alelo
natural o común. Las otras versiones del gen son alelos varian-
tes o mutantes que se diferencian del alelo natural debido a la
presencia de una mutación, es decir, un cambio permanente en
la secuencia de nucleótidos o en la disposición del DNA. Un
conjunto dado de alelos localizados en un locus, o un conjunto
de loci en un cromosoma, se denomina un haplotipo.
Los alelos variantes se originan a partir de mutaciones
que han tenido lugar en el pasado reciente o remoto. Si en un
grupo de población existen al menos dos alelos relativamente
frecuentes en un locus, se dice que este locus presenta polimor-
fi smo (literalmente, «muchas formas»), tal como se expone con
detalle los capítulos siguientes. Además de un alelo normal o
de la existencia de alelos polimorfos comunes, los loci tam-
bién pueden presentar uno o más alelos variantes infrecuentes.
Algunos de estos alelos infrecuentes fueron identifi cados ori-
ginalmente debido a que causan enfermedad genética; otros
pueden incrementar la susceptibilidad frente a la enfermedad
y, fi nalmente, otros carecen de signifi cación conocida en lo que
se refi ere a la salud.
El término mutación se utiliza en genética médica en dos
sentidos; en ocasiones para indicar un nuevo cambio genético
que no se conocía previamente en una familia, y otras veces
simplemente para indicar un alelo mutante que causa una en-
fermedad. Sin embargo, los términos de mutación y mutante
no se utilizan nunca para designar a las personas que son por-
tadoras de alelos mutantes.
Genotipo y fenotipo
El genotipo de una persona es el conjunto de alelos que da
lugar a su constitución genética, tanto de manera conjunta en
todos los loci como –lo más habitual– en un único locus. Por
el contrario, el fenotipo es la expresión observable de un ge-
notipo con sus características morfológicas, clínicas, celulares
y bioquímicas. Habitualmente, se considera que el fenotipo
indica la presencia o la ausencia de una enfermedad, pero en
realidad el fenotipo se puede referir a cualquier manifestación
patológica, incluyendo las características que sólo se pueden
detectar mediante el análisis de la sangre o el estudio de los te-
jidos. Por supuesto, el fenotipo puede ser normal o patológico
en un individuo concreto, pero en este libro (en el que se con-
sideran básicamente los trastornos con signifi cación médica)
el objetivo es el estudio de los fenotipos anómalos, es decir, de
los trastornos genéticos. A pesar de que cada gen codifi ca ge-
neralmente una cadena polipeptídica o una molécula de RNA,
un único gen anómalo o un par de genes anómalos dan lugar
a menudo a efectos fenotípicos múltiples y diversos, y deter-
minan los órganos y sistemas afectados, los signos y síntomas
concretos que se van a producir, y también el momento en el
que van a aparecer. En estas circunstancias, decimos que la
expresión del defecto genético es pleiotrópica. Actualmente,
en lo que se refi ere a muchos trastornos pleiotrópicos, se des-
conoce o no es obvia la conexión entre el defecto genético y las
diversas manifestaciones.
Un trastorno monogénico es el que está determinado
principalmente por los alelos localizados en un único locus.
Cuando una persona posee un par de alelos idénticos en un lo-
cus codifi cado en el DNA nuclear, decimos que es homocigota;
cuando los alelos son diferentes, decimos que es heterocigota
o portadora. El término de heterocigoto compuesto se utiliza
para describir un genotipo en el que están presentes dos alelos
mutantes diferentes del mismo gen, más que un alelo normal
y otro mutante. Estos términos (homocigoto, heterocigoto y
heterocigoto compuesto) se pueden aplicar a una persona o a
un genotipo. En el caso especial en el que una persona de sexo
masculino posee un alelo anómalo de un gen localizado en el
cromosoma X y no existe otra copia del gen, no es homocigota
ni heterocigota, y se denomina hemicigota. El DNA mitocon-
drial también es un caso especial. A diferencia de las dos copias
de cada gen existentes en cada célula diploide, las moléculas
de DNA mitocondrial y los genes codifi cados por el genoma
mitocondrial presentan entre decenas y miles de copias por
célula (v. cap. 2). Por esta razón, los términos de homocigoto,
heterocigoto y hemicigoto no se utilizan para describir los ge-
notipos en los loci mitocondriales.
Árboles genealógicos
Los trastornos monogénicos se caracterizan por sus patrones de
transmisión en las familias. Para establecer el patrón de trans-
misión, un primer paso habitual es la obtención de información
relativa a la historia familiar del paciente; el resumen de los de-
talles de esta información queda plasmado en el árbol genealó-
gico, una representación gráfi ca del árbol familiar en la que se
utilizan símbolos estándar (fi g. 7-1). La familia ampliada que
queda recogida en este tipo de árboles genealógicos se deno-
mina árbol de parentesco (fi g. 7-2). El miembro de la familia a
través del cual el especialista en genética detecta inicialmente la
presencia de un trastorno genético es el probando (sinónimos,
propósito o caso índice) en los casos en los que está afectado. La
persona que se pone en contacto con el especialista en genética
para realizar una consulta sobre su familia es el consultante,
que, a su vez, puede ser un familiar afectado o no afectado del
probando. Una familia puede tener más de un probando si es
valorada a través de más de una fuente. Los probandos tienen
hermanos y hermanas (sibs, en inglés)y el conjunto de ellos se
denomina conjunto de hermanos (sibship, en inglés). Los parien-
tes se clasifi can en familiares de primer grado (padres, hermanos
e hijos del probando), familiares de segundo grado (abuelos y
nietos, tíos y tías, sobrinos y sobrinas, y hermanastros), fami-
liares de tercer grado (p. ej., primos hermanos), etc.., según el
número de pasos que exista en el árbol genealógico entre dos
parientes. Los hijos de primos hermanos son primos segundos,
y un niño es un «primo hermano de primera generación» de
los primos hermanos de sus padres. Las parejas con uno o más
antepasados comunes son consanguíneas. Si solamente existe
un miembro de la familia afectado, hablamos de caso aislado;
en las situaciones en las que el trastorno se debe a una mu-
tación nueva en el probando, hablamos de caso esporádico
(v. fi g. 7-2). Cuando existe una similitud intensa del fenotipo
entre familias diferentes que presentan el mismo defecto, a me-
CAPÍTULO 7
●
Patrones de herencia monogénica
117
© Elsevier. Es una publicación MASSON. Fotocopiar sin autorización es un delito.
Individuo de sexo masculino
Matrimonio
o emparejamiento
Aborto espontáneo
Emparejamientos
múltiples
Embarazo con
información sobre
fechas, si fuera
posible
Gemelos
monocigóticos
Sin descendencia
Gemelos
dicigóticos
Gemelos de
cigosidad
desconocida
Consanguinidad
Divorcio
Individuo fallecido
Individuo adoptado
por la familia evaluada
Familiar adoptado
por otra familia distinta
Aborto
espontáneo
Interrupción del
embarazo
La flecha indica el
consultante que
solicita el consejo
genético
Aborto
Probando
Sexo no especificado
Individuo de sexo femenino
Número de hijos
del sexo indicado
Afectados
Portador sin penetrancia,
puede manifestar la enfermedad
Portador obligado,
no manifiesta la enfermedad
P
?
Árbol genealógico
con numeración de
las generaciones y
los individuos
P
II
I
12
1
2
123
32
P
24 wk
P
LMP
12/20/05
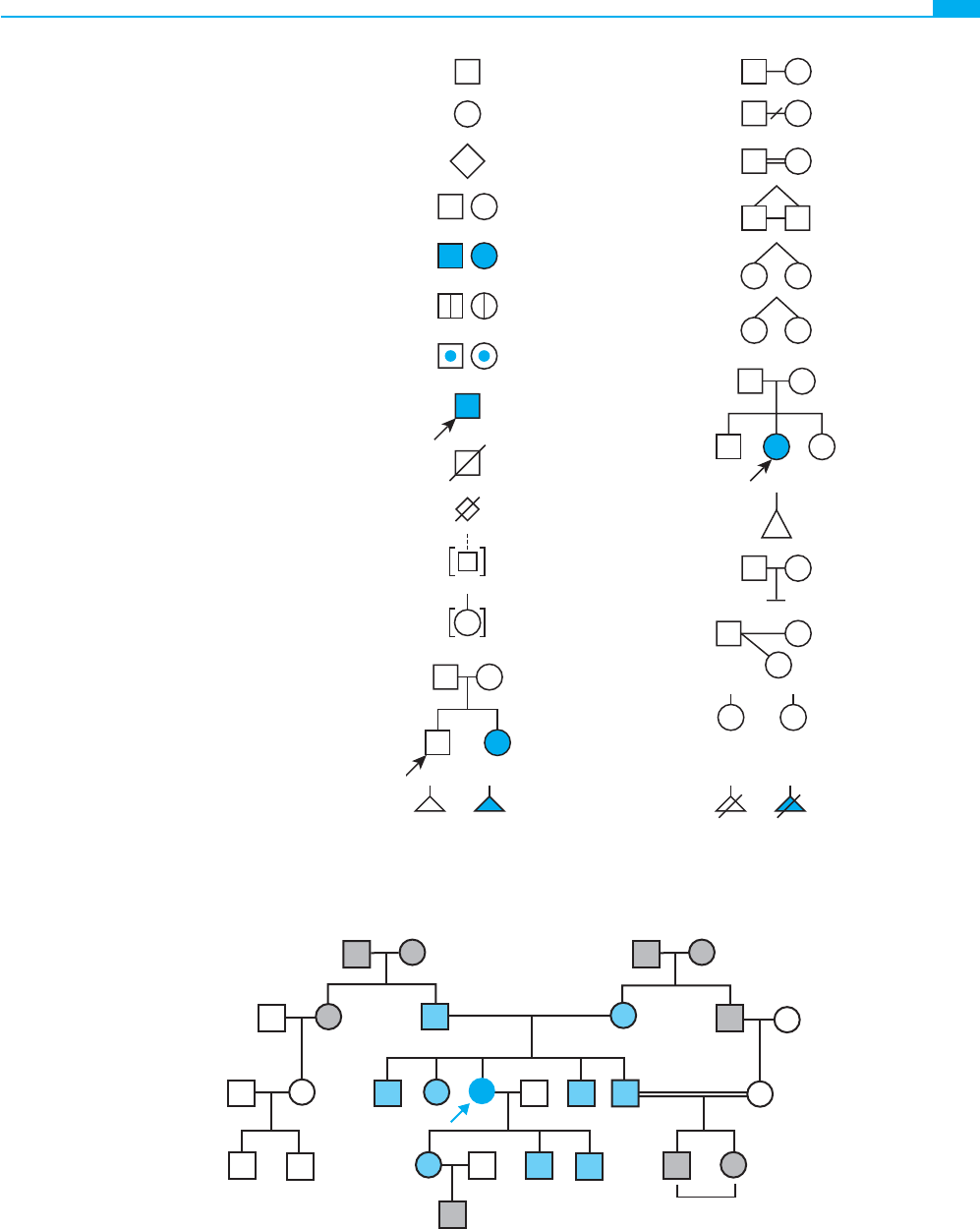
Figura 7-1 ■ Símbolos utilizados habitualmente
en las representaciones gráfi cas de los árboles genea-
lógicos. A pesar de que no hay un sistema uniforme
de notación en este tipo de gráfi cos, los símbolos que
se presentan en la fi gura están fundamentados en las
recomendaciones recientes efectuadas por los profe-
sionales en el campo del consejo genético. (Tomada
de Bennett RL, Steinhaus KA, Uhrich SB et al:
Recommendations for standardized pedigree nomen-
clature. J Genet Counsel 4:267-279, 1995.)
I
II
III
IV
V
12 34
654321
12 345678 9
1
87654321
2°
2°
2°
2°
2°
2°
1°
1°
1°3°
1°
1°
1° 3°
2°
4°
4°
1°
1°
1°
2° y 4°
Figura 7-2 ■ Relaciones en un árbol familiar. La probando, III-5 (fl echa), representa un caso aislado de un trastorno genético.
Tiene cuatro hermanos, III-3, III-4, III-7 y III-8. Su pareja/cónyuge es III-6, y tienen tres hijos (su progenie F1). La probando
tiene nueve familiares en primer grado (sus padres, sus hermanos y sus hijos), nueve familiares en segundo grado (abuelos, tíos
y tías, sobrinos y sobrinas, y nietos), dos familiares en tercer grado (primos hermanos) y cuatro familiares en cuarto grado (primo
hermano de primera generación). IV-3, IV-5 y IV-6 son primos segundos de IV-1 y IV-2. IV-7 y IV-8, cuyos padres son consan-
guíneos, están doblemente relacionados con la probando: son familiares en segundo grado a través del padre y familiares en
cuarto grado a través de la madre.

Thompson & Thompson GENÉTICA EN MEDICINA
118
nudo es posible utilizar patrones de herencia bien establecidos
en otras familias que sufren el mismo trastorno con objeto de
establecer el diagnóstico y de realizar el consejo genético, a pe-
sar de que el paciente constituya un caso aislado en la familia.
Así, muchos pacientes con trastornos genéticos no presentan
similitud con familiares afectados, aunque todavía es posible
reconocer que el trastorno tiene un origen genético.
En lo relativo a muchos trastornos, el hecho de que el
problema presente o no un patrón familiar obvio de trans-
misión en las familias depende de la posibilidad de que los
individuos afectados se puedan reproducir. Los especialistas
en genética han acuñado el término de capacidad reproduc-
tiva para determinar el impacto de un trastorno sobre la
reproducción. La capacidad reproductiva se defi ne como el
número de individuos de la descendencia afectados por el
problema y que puede sobrevivir hasta su edad reproductiva,
en comparación con un grupo control apropiado. La capaci-
dad reproductiva no es una medida de la discapacidad física
o mental. Por ejemplo, en algunos trastornos los pacientes
pueden presentar una capacidad mental y un estado de salud
normales, a pesar de lo cual muestran una capacidad repro-
ductiva de 0 debido a que el problema interfi ere con su repro-
ducción. En otros casos, un trastornos genético grave puede
acompañarse de una capacidad reproductiva normal debido
a que la enfermedad se inicia mucho tiempo después de la
edad reproductiva habitual.
HERENCIA MENDELIANA
Los patrones que muestran los trastornos monogénicos en los
árboles genealógicos dependen principalmente de dos facto-
res:
1. El hecho de que el fenotipo es dominante (se expresa sola-
mente cuando uno de un par de cromosomas es portador
del alelo mutante y el otro cromosoma presenta un alelo
natural en el locus correspondiente) o recesivo (se expresa
solamente cuando ambos cromosomas son portadores de
un par de alelos mutantes en un locus).
2. La localización cromosómica del locus del gen, que puede
ser un autosoma (cromosomas 1 a 22) o un cromosoma
sexual (cromosomas X e Y).
No obstante, es necesario diferenciar entre los genes que se
localizan físicamente en los cromosomas sexuales (sintenia X
o Y) y los genes que muestran una herencia ligada al cromo-
soma X (o ligada al cromosoma Y). La mayoría de los loci
existentes en el cromosoma X muestra una transmisión here-
ditaria ligada a este cromosoma debido a que participa en la
recombinación meiótica únicamente durante la gametogénesis
femenina, cuando existen dos cromosomas X, pero no mues-
tra recombinación con el cromosoma Y durante la gameto-
génesis masculina. Sin embargo, hay un pequeño número de
genes (denominados loci seudoautosómicos; se exponen más
adelante en este capítulo) localizados en el cromosoma X que
no muestran una transmisión hereditaria ligada al cromosoma
X debido a que se pueden combinar con los genes correspon-
dientes del cromosoma Y. Por tanto, existen cuatro patrones
básicos de herencia monogénica (si agrupamos los patrones
autosómicos y seudoautosómicos):
Dominante Recesiva
Herencia
autosómica Autosómica dominante Autosómica recesiva
Herencia ligada Dominante ligada al Recesiva ligada al
cromosoma X cromosoma X cromosoma X
Además de estos patrones clásicos de árbol genealógico
observados respecto a los alelos causantes de enfermedad lo-
calizados en loci de cromosomas situados en el núcleo, hay
otra clase de trastornos con un patrón materno distintivo de
herencia que pueden ser debidos a mutaciones en el genoma
mitocondrial (se describen más adelante en este capítulo).
Herencias autosómica y ligada al cromosoma X
El hecho de que un gen anómalo esté localizado en un autosoma
o aparezca ligado al cromosoma X infl uye de manera profunda
en la expresión clínica de la enfermedad. En primer lugar, en
general, los trastornos autosómicos afectan por igual a hombres
y mujeres. (Las únicas excepciones son los denominados tras-
tornos con limitación sexual, expuestos más adelante en este
capítulo.) En lo relativo a los trastornos ligados al cromosoma
X, la situación es muy diferente. Los hombres sólo presentan un
cromosoma X y, por tanto, son hemicigotos respecto a los genes
ligados al cromosoma X; los hombres 46,XY nunca son hete-
rocigotos para los alelos de loci localizados en el cromosoma
X, mientras que las mujeres pueden ser heterocigotas u homo-
cigotas respecto a los loci ligados al cromosoma X. En segundo
lugar, para compensar el complemento doble de genes ligados
al cromosoma X en las mujeres, los alelos de la mayor parte de
los genes ligados al cromosoma X se expresa únicamente en uno
de los dos cromosomas X de una célula dada de una mujer (tal
como se describe en el cap. 6).
Herencias dominante y recesiva
Herencia recesiva
Tal como se defi ne clásicamente, es recesivo un fenotipo expre-
sado sólo por los homocigotos (o bien, en lo relativo a los ras-
gos ligados a X, por los hemicigotos de sexo masculino) y no
por los heterocigotos. La mayor parte de los trastornos rece-
sivos descritos hasta el momento se debe a mutaciones que re-
ducen o eliminan la función del producto del gen, en lo que se
denomina mutaciones con pérdida de función. Por ejemplo,
muchas enfermedades recesivas se deben a mutaciones que
alteran o eliminan la función de una enzima. Generalmente,
estas mutaciones se heredan en forma de enfermedades rece-
sivas debido a que los heterocigotos, que solamente presentan
un par de alelos funcionales mientras que el otro alelo no lo es
(el alelo patológico), pueden elaborar característicamente una
cantidad sufi ciente del producto (aproximadamente, el 50%
del que producen los homocigotos de tipo natural) para llevar
a cabo la reacción enzimática necesaria para la función fi sioló-
gica normal, evitando así la enfermedad (v. cap. 12).
Herencia dominante
Por el contrario, un fenotipo expresado tanto por los homocigo-
tos como por los heterocigotos para un alelo mutante se hereda
CAPÍTULO 7
●
Patrones de herencia monogénica
119
© Elsevier. Es una publicación MASSON. Fotocopiar sin autorización es un delito.
de manera dominante. Los trastornos dominantes aparecen tan-
to si el alelo normal restante da lugar a la producción normal
de un gen como si no es así. En las enfermedades dominantes
puras están afectados de manera similar los homocigotos y los
heterocigotos respecto al alelo mutante. Realmente, en genética
médica los trastornos dominantes puros son escasos o incluso
inexistentes. En ocasiones tiene lugar la expresión fenotípica
de dos alelos diferentes respecto a un locus, en cuyo caso los
dos alelos se denominan codominantes. Un ejemplo bien co-
nocido de expresión codominante es el del sistema de grupos
sanguíneos ABO (v. cap. 9). Lo más habitual es que los tras-
tornos dominantes sean más graves en los homocigotos que en
los heterocigotos, en cuyo caso decimos que la enfermedad es
dominante incompleta (o semidominante). Los diferentes meca-
nismos moleculares que explican las razones por las que ciertas
mutaciones dan lugar a una enfermedad hereditaria dominante
y no recesiva se exponen en el capítulo 12.
En términos estrictos, es la herencia de un fenotipo (más
que la de un alelo) lo que es dominante o recesivo. Sin embargo,
los alelos mutantes se denominan a menudo dominantes o rece-
sivos según si pueden modifi car el fenotipo en los estados hete-
rocigotos u homocigotos, respectivamente. En consecuencia, los
términos de alelo o gen dominante y de alelo o gen recesivo se
utilizan con frecuencia, si bien de manera un poco laxa.
FACTORES QUE INFLUYEN EN LOS
PATRONES DE LOS ÁRBOLES
GENEALÓGICOS
Penetrancia y expresividad
Muchos trastornos genéticos presentan una segregación bien
defi nida en las familias, lo que quiere decir que el fenotipo
anómalo se puede diferenciar fácilmente del fenotipo normal.
Sin embargo, en la experiencia clínica algunos trastornos no
se expresan en el absoluto en un individuo a pesar de que este
individuo presente el mismo genotipo que da lugar a la expre-
sión de la enfermedad en otros miembros de su familia. En
estos otros casos, el mismo trastorno puede manifestar una
expresión extremadamente variable en términos de gravedad
clínica, de la gama de síntomas o de la edad de inicio. La ex-
presión fenotípica de un genotipo anómalo se puede modifi car
por los efectos del envejecimiento, de otros loci genéticos o de
factores ambientales, y estas diferencias en la expresión pue-
den dar lugar con frecuencia a difi cultades en el diagnóstico y
en la interpretación del árbol genealógico. Hay dos mecanis-
mos bien defi nidos a través de los cuales se pueden producir
estas diferencias en la expresión: la penetrancia reducida y la
expresividad variable.
La penetrancia es la probabilidad de que un gen presente
cualquier nivel de expresión fenotípica. Cuando la frecuen-
cia de expresión de un fenotipo es inferior al 100% (es decir,
cuando algunos de los individuos con el genotipo apropiado
no muestran en absoluto el fenotipo correspondiente), decimos
que el gen muestra una penetrancia reducida. La penetrancia
es un concepto de todo o nada. Es el porcentaje de personas
con un genotipo de predisposición que sufre realmente la en-
fermedad, al menos en un cierto grado.
La expresividad es la gravedad de la expresión del fenoti-
po en individuos que presentan el mismo genotipo causante de
la enfermedad. Cuando la gravedad de la enfermedad difi ere
en las personas que poseen el mismo genotipo, decimos que
el fenotipo muestra una expresividad variable. Incluso en el
mismo árbol de parentesco, dos individuos portadores de los
mismos genes mutantes pueden presentar algunos signos y sín-
tomas en común, mientras que el resto de las manifestaciones
de sus enfermedades respectivas puede ser muy diferente, se-
gún los tejidos u órganos afectados.
Algunas de las difi cultades que acompañan a la penetran-
cia dependiente de la edad y a la expresividad variable respec-
to al conocimiento de la herencia de un fenotipo patológico
quedan demostradas en la enfermedad autosómica dominante
neurofi bromatosis (NF1)
(Caso 29) . La NF1 es un trastorno
frecuente del sistema nervioso, los ojos y la piel que se observa
en aproximadamente uno de cada 3.500 recién nacidos. No
existe una variación signifi cativa en la frecuencia de la enfer-
medad entre los distintos grupos raciales. En la fi gura 7-3 se
muestra una de las manifestaciones clínicas características. La
NF1 se caracteriza por la aparición de múltiples tumores be-
nignos de consistencia carnosa (los neurofi bromas) en la piel;
de múltiples lesiones cutáneas planas, irregulares y pigmen-
tadas denominadas «máculas café con leche»; de pequeños
tumores benignos (hamartomas) en el iris ocular denomina-
dos nódulos de Lisch, y de otros problemas menos frecuentes
como retraso mental, tumores en el sistema nervioso central,
neurofi bromas plexiformes y tumores malignos en el sistema
nervioso periférico o el músculo esquelético. Por tanto, este
trastorno da lugar a un fenotipo pleiotrópico.
La NF1 fue descrita con detalle por el médico von Rec-
klinghausen en 1882, aunque posiblemente esta enfermedad
ya se conocía desde la antigüedad. A pesar de que los hetero-
cigotos adultos muestran casi siempre algún signo de la enfer-
medad (por tanto, su penetrancia es del 100% en los adultos),
algunos de ellos presentan solamente máculas café con leche,
pecas en la piel axilar y nódulos de Lisch, mientras que otros
pueden presentar tumores benignos (aunque potencialmente
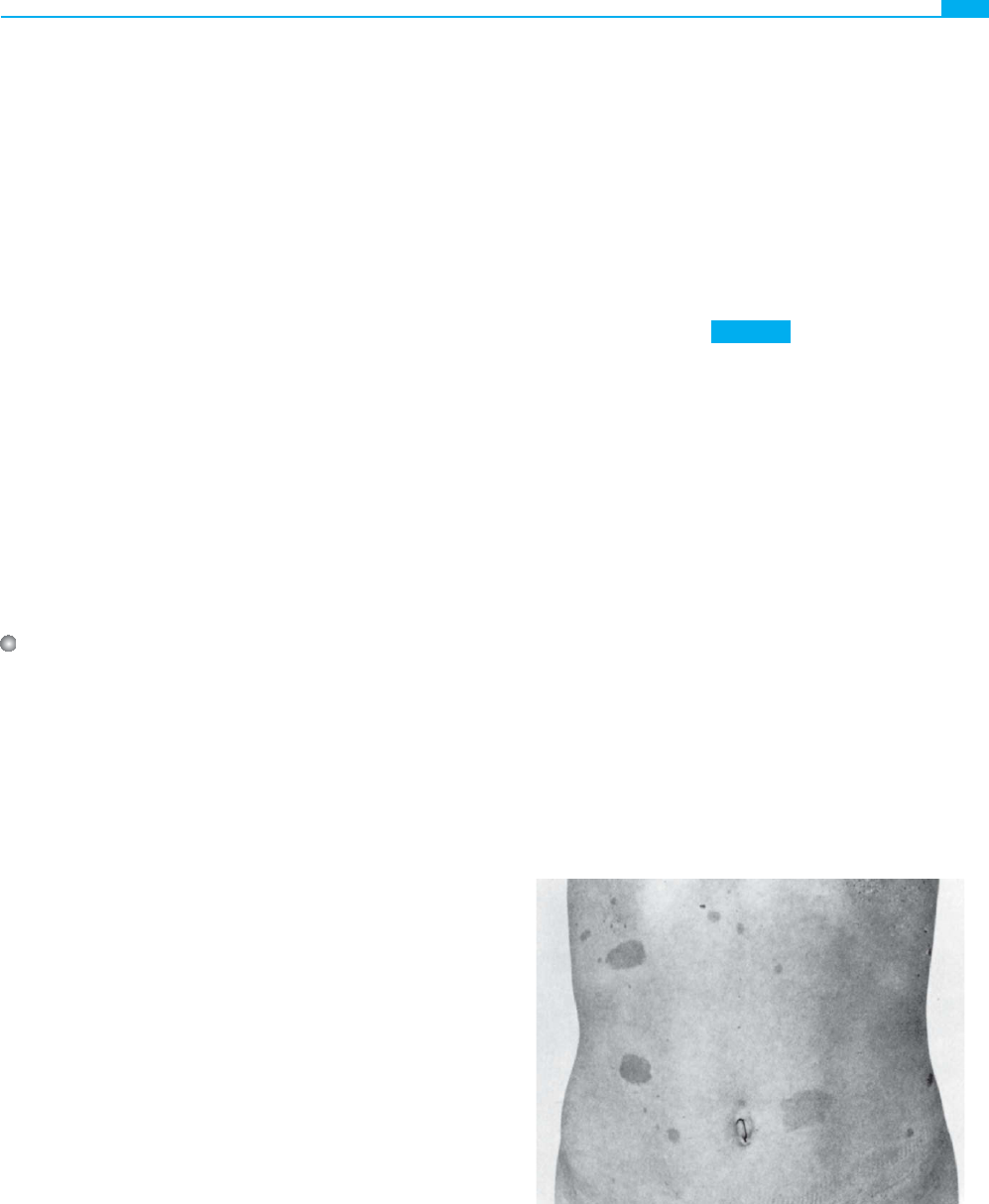
Figura 7-3 ■ Neurofi bromatosis tipo 1: máculas cutáneas
«café con leche» hiperpigmentadas, que constituyen un signo
diagnóstico útil en los familiares que, por lo demás, podrían
ser considerados no afectados. La mayor parte de los pacientes
presenta seis o más de estas máculas con un diámetro de al
menos 15 mm, generalmente en el tronco. (Cortesía de
Rosanna Weksberg, The Hospital for Sick Children, Toronto,
Canadá.)
Thompson & Thompson GENÉTICA EN MEDICINA
120
mortales) que afectan a la médula espinal o bien sarcomas de
alto grado de malignidad en las extremidades. Así, la expre-
sividad es variable; incluso dentro de un grupo de hermanos,
algunos individuos presentan una afectación grave mientras
que otros muestran solamente una afectación leve. El diagnós-
tico se complica aún más en los niños debido a que los signos
de la enfermedad se desarrollan de manera gradual durante la
niñez. Por ejemplo, durante el periodo de recién nacido menos
de la mitad de los recién nacidos afectados muestra incluso el
signo más sutil de la enfermedad, es decir, el incremento en el
número de máculas café con leche. Por tanto, la penetrancia
depende de la edad.
En el gen NF1 se han observado muchas mutaciones di-
ferentes, todas los cuales parecen dar lugar a la pérdida de
función del producto de este gen, la neurofi bromina. Aproxi-
madamente, la mitad de los pacientes con NF1 sufre la enfer-
medad debido a una mutación nueva, más que a una mutación
heredada (fi g. 7-4).
El principal problema que se plantea en el consejo ge-
nético ofrecido a las familias con NF1 es el de decidir entre
dos posibilidades que muestran una probabilidad similar: ¿la
enfermedad en el probando es esporádica (es decir, se debe
a una mutación nueva) o bien el probando ha heredado una
forma clínicamente signifi cativa de la enfermedad a partir de
un progenitor que presenta el gen pero que sólo muestra una
expresión leve del mismo? Si el probando ha heredado el de-
fecto genético, el riesgo de que sus hijos también lo hereden
es del 50%; sin embargo, si la enfermedad del probando es
el resultado de una mutación genética nueva, el riesgo de que
cualquiera de sus hijos sufra la enfermedad es muy bajo. Un
aspecto signifi cativo es que, en los dos casos, el riesgo de que
el paciente pueda transmitir el gen a cualquiera de sus hijos
es del 50%. A pesar de estas incertidumbres, las familias de
los pacientes con NF1 se suelen sentir tranquilizadas cuando
saben que es posible la detección presintomática de la enfer-
medad y que incluso el trastorno se puede determinar antes del
nacimiento mediante análisis genético molecular (v. cap. 17).
Por desgracia, el análisis molecular solamente puede indicar en
la mayor parte de los casos si el trastorno va a manifestarse,
pero no la gravedad con la que lo va a hacer. Excepto en lo que
se refi ere a la asociación de deleciones de genes completos con
características dismórfi cas, retraso mental y un elevado núme-
ro de neurofi bromas a edades tempranas, no existe ninguna
correlación entre la gravedad del fenotipo y los alelos NF1
mutantes concretos.
Otro ejemplo de una malformación autosómica dominan-
te con penetrancia reducida es la deformidad en mano hendi-
da, un tipo de ectrodactilia (fi g. 7-5). Esta malformación se
origina a la sexta o séptima semanas del desarrollo, en la épo-
ca de formación de las manos y los pies. El trastorno muestra
heterogeneidad de locus y se han reconocido al menos cinco
loci, aunque el gen real responsable del trastorno sólo se ha
identifi cado en unos pocos casos. La falta de penetrancia en
los árboles genealógicos de la malformación de la mano hen-
dida puede dar lugar a un salto aparente de generaciones, lo
que complica el consejo genético debido a que las personas
con riesgo y manos normales pueden ser, a pesar de ello, por-
tadoras del gen de la enfermedad y por tanto pueden tener
hijos afectados.
La fi gura 7-6 es un árbol genealógico de la deformidad de
la mano hendida en el que la persona no afectada es la consul-
tante (la persona que solicita el consejo genético). Su madre es
portadora de la mutación de la mano hendida, sin penetrancia.
La revisión de la bibliografía correspondiente a la deformidad
de la mano hendida sugiere que existe una penetrancia redu-
cida de aproximadamente el 70% (es decir, solamente el 70%
de las personas portadoras del gen sufre el defecto). La utiliza-
ción de esta información en el análisis bayesiano, un método
matemático para determinar las probabilidades condicionadas
en los árboles genealógicos (se expone con mayor detalle en el
cap. 19) permite calcular el riesgo de que el consultante pueda
tener un hijo con la alteración.
Edad de inicio
Los trastornos genéticos pueden aparecer en cualquier época
de la vida de un individuo, desde las fases tempranas del desa-
rrollo intrauterino hasta los años posteriores a la pérdida de la
capacidad reproductiva. Algunos pueden ser letales antes del
nacimiento, mientras que otros pueden interferir con el desa-
rrollo fetal normal y pueden ser diagnosticados antes de que
tenga lugar el nacimiento (p. ej., mediante ecografía; v. cap.
15), aunque son compatibles con el nacimiento de un niño
vivo; otros trastornos genéticos solamente se pueden recono-
cer tras el nacimiento (congénitos). (Los términos genético y
congénito se confunden entre sí con frecuencia. Hay que te-
ner en cuenta que un trastorno genético está determinado por
los genes, mientras que el trastorno congénito es simplemente
aquel que se detecta en el momento del nacimiento y que puede
tener o no una causa genética.) Por tanto, en un árbol familiar
con una enfermedad letal que afecta a un feto en las primeras
fases del embarazo, el patrón de la enfermedad puede no estar
claro debido a que todo lo que uno observa pueden ser abortos
múltiples o una disminución aparente de la fertilidad, más que
la recurrencia de la enfermedad prenatal en sí misma. Por el
contrario, en una familia en la que existe un trastorno domi-
nante de inicio en edades tardías, es posible que los padres y
los hijos de un individuo afectado no muestren la enfermedad
debido a que el progenitor portador falleció por causas no re-
lacionadas antes de que desarrollara la enfermedad y debido
también a que los niños en riesgo todavía no han alcanzado la
edad a la que se manifi esta el defecto del gen mutante a través
del fenotipo correspondiente a la propia enfermedad.
I
II
III
IV
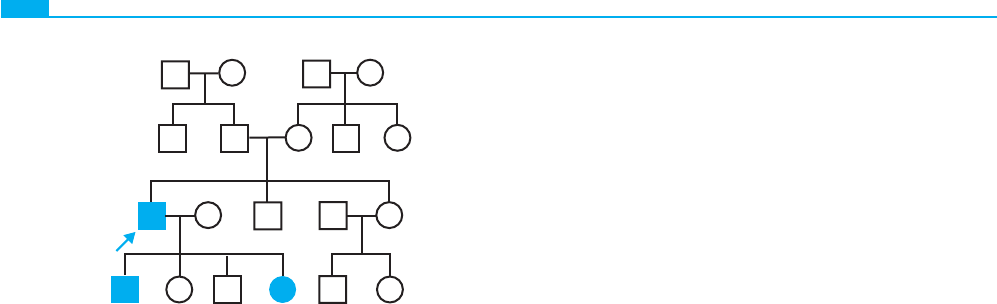
Figura 7-4 ■ Árbol de una familia con neurofi bromatosis
tipo 1, que orienta aparentemente hacia una nueva mutación
en el probando de la generación III (fl echa). Este individuo
parece presentar un alelo de NF1 correspondiente a una muta-
ción nueva, debido a que sus padres y los padres de sus padres
no están afectados.
CAPÍTULO 7
●
Patrones de herencia monogénica
121
© Elsevier. Es una publicación MASSON. Fotocopiar sin autorización es un delito.
Otros factores que infl uyen en los patrones
de los árboles genealógicos
A pesar de que, como norma general, los árboles genealógicos
de los trastornos monogénicos se pueden clasifi car fácilmente
como autosómicos o ligados al cromosoma X, y como domi-
nantes o recesivos, el patrón hereditario de un árbol genealógi-
co individual puede estar oscurecido por algunos otros facto-
res que difi cultan la interpretación de la forma de transmisión
hereditaria. Las difi cultades diagnósticas pueden ser debidas a
una penetrancia reducida o a una expresividad variable de la
enfermedad; a la posibilidad de que haya otros genes y facto-
res ambientales que infl uyen en la expresión genética; al hecho
de que los individuos con ciertos fenotipos no sobreviven has-
ta el momento del nacimiento; a la inexistencia de información
precisa respecto a la presencia del trastorno en los familiares,
o respecto a las propias relaciones familiares; a la aparición de
mutaciones nuevas que pueden contribuir a que la enferme-
dad sea dominante y ligada al cromosoma X, y, fi nalmente, al
hecho de que el tamaño característico de las familias hoy día
es pequeño en la mayor parte de los países desarrollados, de
manera que el paciente puede ser por azar el único miembro de
la familia afectado, lo que hace muy difícil la determinación de
cualquier patrón de herencia.
CORRELACIÓN ENTRE GENOTIPO
Y FENOTIPO
Un componente importante de la genética médica es la iden-
tifi cación y caracterización de los genotipos responsables de
los fenotipos concretos de la enfermedad. Es importante no
adoptar un punto de vista demasiado simplista respecto a la
relación existente entre las mutaciones monogénicas y los fe-
notipos de la enfermedad. Cuando se analiza con detalle un
trastorno genético que aparentemente es hereditario en forma
de un trastorno monogénico, a menudo se observa que es ge-
néticamente heterogéneo; es decir, que incluye varios fenotipos
que son similares pero que están determinados realmente por
genotipos diferentes localizados en loci distintos. La heteroge-
neidad genética puede ser el resultado de la presencia de mu-
taciones diferentes en el mismo locus (heterogeneidad alélica),
de mutaciones en loci diferentes (heterogeneidad de locus) o
de ambas posibilidades (v. cap. 12). El reconocimiento de la
heterogeneidad genética es un aspecto importante del diagnós-
A
B
Figura 7-5
■ Deformidad de la mano hendida, un trastorno autosómico dominante que afecta a las manos y los pies, en un
niño de 3 meses de edad. A: Parte superior del cuerpo. B: Parte inferior del cuerpo. (Tomada de Kelikian H: Congenital Defor-
mities of the Hand and Forearm. Filadelfi a, WB Saunders, 1974.)
III
II
I
Figura 7-6 ■ Árbol de una familia con la deformidad de
la mano hendida, en la que se observa la falta de penetrancia
en la madre de la consultante (fl echa). La penetrancia reducida
se debe tener en cuenta en el consejo genético.
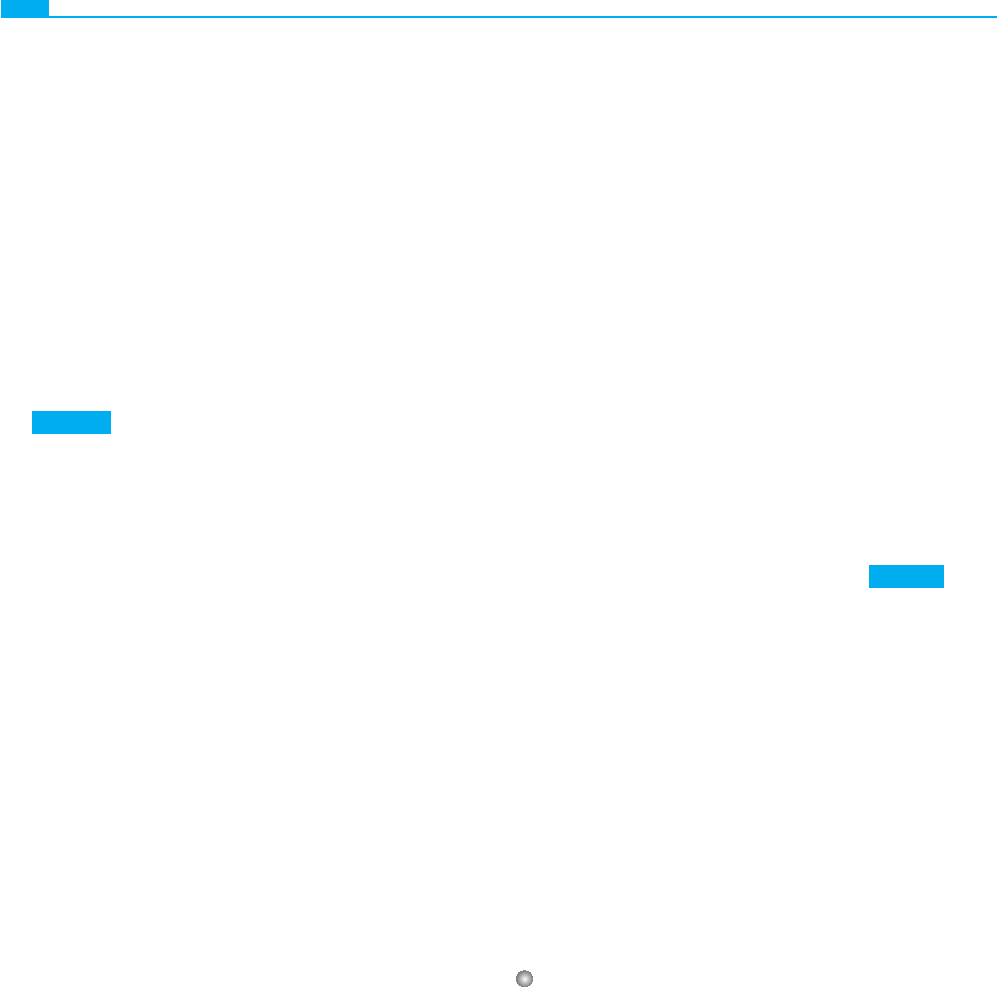
Thompson & Thompson GENÉTICA EN MEDICINA
122
tico clínico y del consejo genético. Por otra parte, los fenotipos
bien defi nidos heredados en las diferentes familias pueden ser
debidos a alelos mutantes diferentes correspondientes al mis-
mo gen. Este fenómeno, denominado heterogeneidad clínica o
fenotípica, es bien conocido y se debe tener en cuenta a la hora
de establecer correlaciones entre el genotipo y el fenotipo.
Heterogeneidad alélica
La heterogeneidad alélica es una causa importante de la va-
riación clínica. Muchos loci poseen más de un alelo mutante;
de hecho, en un locus dado puede haber varias mutaciones o
mutaciones múltiples. Por ejemplo, en todo el mundo se han
observado casi 1.400 mutaciones distintas en el regulador de
la conductancia transmembrana de la fi brosis quística (CFTR,
cystic fi brosis transmembrane conductance regulator; v. cap.
12)
(Caso 10) . En ocasiones, estas diferentes mutaciones dan
lugar a trastornos clínicamente indistinguibles. En otros casos,
los alelos mutantes diferentes correspondientes al mismo locus
dan lugar a un fenotipo similar pero con un espectro de gra-
vedad; por ejemplo, algunas mutaciones CFTR pueden hacer
que los pacientes sufran la forma clásica de la fi brosis quística,
con insufi ciencia pancreática, enfermedad pulmonar grave y
progresiva, y ausencia congénita de los conductos deferentes
en los hombres, mientras que los pacientes portadores de otros
alelos mutantes pueden manifestar la afectación pulmonar
pero con una función pancreática normal, y otros pueden pre-
sentar únicamente las alteraciones correspondientes al tracto
reproductor masculino.
Dado que cualquier alelo mutante concreto suele ser infre-
cuente en la población general, la mayor parte de las personas
que sufren trastornos autosómicos recesivos son heterocigotos
compuestos, más que homocigotos verdaderos. Debido a que
las diferentes combinaciones de alelos pueden dar lugar a con-
secuencias clínicas ligeramente diferentes, los clínicos tienen
que tener en cuenta la heterogeneidad alélica como una posi-
ble explicación de la variabilidad en los pacientes que se con-
sidera sufren la misma enfermedad. No obstante, hay algunas
excepciones bien reconocidas a la observación de que los hete-
rocigotos compuestos son más frecuentes que los homocigotos
verdaderos. La primera es la que se refi ere a la situación en la
que los individuos afectados heredan el mismo alelo mutante
a partir de padres consanguíneos que son portadores (los dos)
del mismo alelo mutante heredado a partir de un antepasado
común. La segunda se refi ere a la situación en la que un ale-
lo mutante puede ser responsable de una elevada proporción
de casos de una enfermedad autosómica recesiva en un grupo
racial concreto, de manera que muchos de los pacientes que
constituyen este grupo van a ser homocigotos para dicho ale-
lo. La tercera excepción es la que tiene lugar cuando el tras-
torno muestra normalmente una heterogeneidad alélica esca-
sa o nula debido a que el fenotipo de la enfermedad causada
por una mutación concreta es específi co para dicha mutación
(p. ej., la enfermedad falciforme; v. cap. 11).
(Caso 37).
Heterogeneidad de locus
Con respecto a muchos fenotipos, el análisis del árbol genealó-
gico como evaluación única ha sido sufi ciente para demostrar
la heterogeneidad de locus. Por ejemplo, sabemos desde hace
tiempo que la retinitis pigmentosa (una causa frecuente de al-
teración visual por degeneración de los fotorreceptores) puede
cursar como una enfermedad autosómica dominante, autosó-
mica recesiva o ligada al cromosoma X. Durante los últimos
años se ha determinado que la heterogeneidad es incluso más
intensa; el análisis de los árboles genealógicos, en combinación
con los métodos de cartografía de genes, ha demostrado que
existen al menos 43 loci responsables de cinco formas ligadas
al cromosoma X, de 14 formas autosómicas dominantes y de
24 formas autosómicas recesivas de retinitis pigmentosa que
no se asocian a otras alteraciones fenotípicas. Si consideramos
los trastornos en los que la retinitis pigmentosa aparece junto
a otros defectos como retraso mental o sordera, existen casi
70 enfermedades genéticas que cursan con retinitis pigmentosa.
Heterogeneidad fenotípica
Las mutaciones diferentes en un mismo gen pueden dar lugar en
ocasiones a fenotipos sorprendentemente diferentes. Por ejem-
plo, algunas mutaciones con pérdida de función en el gen RET,
que codifi ca un receptor tipo tirosincinasa, pueden dar lugar a
una falta de desarrollo de los ganglios nerviosos colónicos que
se transmite de manera dominante, con aparición de un tras-
torno de la motilidad colónica y de un estreñimiento crónico y
grave (enfermedad de Hirschsprung; v. cap. 8)
(Caso 20) . Otras
mutaciones en el mismo gen pueden dar lugar a una función
excesiva y no regulada de la cinasa, con aparición de tumores
malignos de herencia dominante en las glándulas tiroides y su-
prarrenales (neoplasia endocrina múltiple tipos 2A y 2B; v. cap.
16). Un tercer grupo de mutaciones en el gen RET causa en el
mismo individuo tanto la enfermedad de Hirschsprung como
la neoplasia endocrina múltiple. Hay una situación comparable
respecto al gen LMNA, que codifi ca las lamininas A/C (proteí-
nas de la membrana nuclear). Las diferentes mutaciones LMNA
se han asociado a media docena de trastornos fenotípicamente
diferentes, tal como la distrofi a muscular de Emery-Dreifuss,
una forma de miocardiopatía dilatada hereditaria, una de las
formas de la neuropatía periférica de Charcot-Marie-Tooth, un
trastorno del tejido adiposo normal denominado lipodistrofi a y
el síndrome de envejecimiento prematuro denominado progeria
de Hutchinson-Gifford.
PATRONES AUTOSÓMICOS DE HERENCIA
MENDELIANA
Herencia autosómica recesiva
Las enfermedades autosómicas recesivas solamente afectan a
los homocigotos y a los heterocigotos compuestos, que son
personas con dos alelos mutantes y que no presentan ningún
alelo normal, debido a que en estas enfermedades una copia
del gen normal puede compensar el alelo mutante e impedir
la aparición del proceso patológico. Dado que un individuo
solamente hereda uno de los dos alelos de cualquier locus a
partir de uno de sus progenitores, los homocigotos deben ha-
ber heredado un alelo mutante de cada uno de sus progenito-
res (a menos que se hayan producido una disomía uniparental
o una mutación nueva, lo que es infrecuente en los trastornos
autosómicos recesivos).
Hay tres tipos de emparejamientos que pueden hacer que
los descendientes sean homocigotos y sufran una enfermedad
autosómica recesiva. El alelo recesivo mutante se indica con el
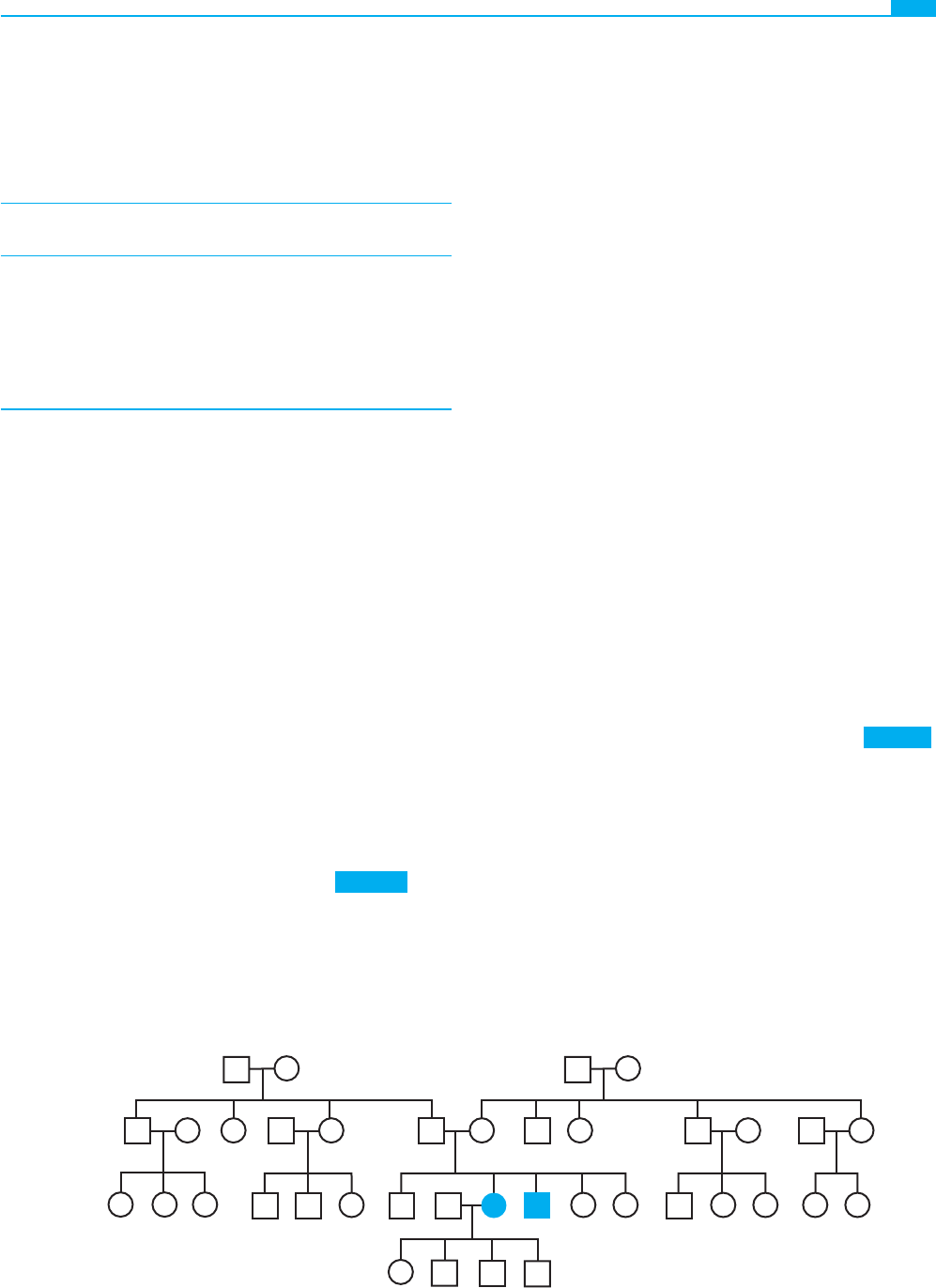
CAPÍTULO 7
●
Patrones de herencia monogénica
123
© Elsevier. Es una publicación MASSON. Fotocopiar sin autorización es un delito.
símbolo de R y el alelo dominante normal con el símbolo de
r. A pesar de que cualquier emparejamiento en el que ambas
personas presentan al menos un alelo recesivo puede dar lugar
a descendientes homocigotos y afectados, el emparejamiento
más frecuente, con diferencia, es el que se produce entre dos
heterocigotos no afectados.
Riesgo de
Emparejamientos Descendientes enfermedad
Portador con portador, 1/4 R/R, 1/2 R/r, 3/4 no afectados,
R/r × R/r 1/4 r/r 1/4 afectados
Portador con afectado, 1/2 R/r, 1/2 r/r 1/2 no afectados,
R/r × r/r 1/2 afectados
Afectado con afectado, r/r solamente Todos afectados
r/r × r/r
Cuando los dos progenitores de una persona afectada son
heterocigotos (portadores), el riesgo de que sus hijos reciban
un alelo recesivo es la mitad respecto a cada uno de los pro-
genitores, de manera que la probabilidad de herencia de dos
alelos recesivos (y, por tanto, de que el hijo esté afectado) es
1/2 x 1/2, es decir, 1 de cada 4. El probando puede ser el único
familiar afectado, pero si hubiera otros familiares que presen-
taran la enfermedad generalmente pertenecerían al mismo gru-
po de hermanos y la enfermedad no afectaría otros segmentos
de la familia (fi g. 7-7).
Trastornos infl uidos por el sexo
Dado que los hombres y las mujeres presentan la misma do-
tación de autosomas, los trastornos autosómicos recesivos
muestran generalmente la misma frecuencia e intensidad en
hombres y mujeres. Sin embargo, hay excepciones. Algunos
fenotipos autosómicos recesivos están infl uidos por el sexo,
lo que quiere decir que se expresan en ambos sexos pero con
frecuencias o gravedades distintas. Entre los trastornos auto-
sómicos dominantes, la hemocromatosis es un ejemplo de un
fenotipo que es más frecuente en los hombres
(Caso 17) . Este
trastorno autosómico recesivo del metabolismo del hierro es
más frecuente en el aproximadamente 0,5% de individuos de
origen nórdico europeo que son homocigotos para una mu-
tación con cambio de sentido que sustituye la cisteína en la
posición 282 por una tirosina (Cys282Tyr) en el gen HFE. Los
homocigotos Cys282Tyr muestran un aumento en la absor-
ción del hierro de la dieta y a menudo presentan alteraciones
analíticas sugestivas de la existencia de reservas corporales de
hierro excesivas, aunque no es frecuente que este trastorno
cause una sobrecarga de hierro con lesiones graves del cora-
zón, el hígado y el páncreas. La menor incidencia del trastor-
no clínico en las mujeres (la quinta a la décima parte que los
hombres) parece estar relacionada, entre otros factores, con el
menor consumo alimentario de hierro, el consumo menor de
alcohol y el aumento en las pérdidas de hierro a través de la
menstruación.
Frecuencia génica y frecuencia de portador
Los alelos mutantes responsables de un trastorno recesivo
son generalmente poco frecuentes, de manera que la mayor
parte de las personas no presenta siquiera una copia del alelo
mutante. Sin embargo, entre los individuos portadores de al
menos una copia del alelo mutante, la frecuencia de los he-
terocigotos clínicamente no afectados poseedores de un alelo
normal y de un alelo mutante siempre es mucho mayor que
la frecuencia de los individuos afectados que son portadores
de dos alelos mutantes infrecuentes. (En el cap. 9 se expone
la manera de calcular las frecuencias reales de los portadores
y de las personas que sufren la enfermedad.) Debido a que un
trastorno autosómico recesivo debe ser heredado a partir de
ambos progenitores, el riesgo de que un portador tenga un
hijo afectado va a depender parcialmente de la posibilidad de
que el otro progenitor del niño también sea portador de un
alelo mutante para la enfermedad. Así, el conocimiento de la
frecuencia del estado de portador en una enfermedad tiene im-
portancia clínica para el consejo genético.
El trastorno autosómico recesivo más frecuente en los ni-
ños de raza blanca es la fi brosis quística (CF, cystic fi brosis),
causada por mutaciones en el gen CFTR (v. cap. 12)
(Caso 10).
La CF es una enfermedad prácticamente desconocida en Asia
y es relativamente rara en las personas de origen afroameri-
cano, pero en los grupos de raza blanca alrededor de uno de
cada 2.000 niños es portador de dos alelos CFTR mutantes y
padece la enfermedad. La frecuencia de portadores de alguno
de los cientos de posibles alelos CFTR mutantes es de aproxi-
madamente 1/29 (v. cap. 9). Por tanto, en una población de
3.247 individuos de raza blanca se puede esperar la existencia
de un paciente con CF, de 112 portadores de una mutación
CFTR no afectados y de 3.134 homocigotos normales. Debido
a que un paciente posee dos alelos CFTR mutantes y a que un
portador sólo presenta uno de estos alelos (112 x 1)/(112 x 1
+ 1 x 2) = 112/114 (aproximadamente, el 98%) de todos los
I
II
III
IV
Figura 7-7 ■ Árbol familiar típico de una herencia autosómica recesiva.

Thompson & Thompson GENÉTICA EN MEDICINA
124
alelos CFTR mutantes en esta población de 3.247 individuos
quedan ocultos en el grupo de portadores (que generalmente
no saben que son portadores), mientras que en los pacientes
sólo se observa el 2% de los alelos mutantes.
Consanguinidad
Dado que la mayor parte de los alelos mutantes responsables de
trastornos autosómicos recesivos afecta a portadores más que a
homocigotos, los alelos mutantes pueden ser transmitidos he-
reditariamente en las familias durante numerosas generaciones
sin que aparezcan individuos homocigotos que sufran la enfer-
medad fl orida. La presencia de estos genes recesivos ocultos no
se revela a menos que ocurra que el portador presente empare-
jamiento con alguna persona que también sea portadora de un
alelo mutante en el mismo locus y que un hijo de ambos herede
los dos alelos de la enfermedad. Se considera que cualquier per-
sona es portadora de al menos ocho a 10 alelos mutantes, de
los cuales quizá la mitad son letales antes del nacimiento en los
homocigotos. El resto da lugar a la aparición de trastornos au-
tosómicos recesivos bien conocidos y fácilmente diagnosticables
en los homocigotos. No obstante, ésta es una estimación míni-
ma que no tiene en cuenta los alelos mutantes que ejercen sus
efectos mediante la interacción con alelos mutantes en otros loci
(herencia multifactorial; v. cap. 8).
La posibilidad de que los dos progenitores sean portado-
res de un alelo mutante localizado en el mismo locus aumenta
de forma considerable si los progenitores están relacionados
genéticamente entre sí, de manera que cada uno de ellos po-
dría haber heredado el alelo mutante a partir de un antepasa-
do común, una situación que se denomina consanguinidad.
La consanguinidad se defi ne arbitrariamente como el empa-
rejamiento de individuos que muestran entre sí una relación
genética más estrecha que la que existe entre los primos segun-
dos. La consanguinidad de los padres de un paciente que sufre
un trastorno genético es una evidencia sólida (aunque no una
prueba) de la herencia autosómica recesiva de dicho trastorno.
Por ejemplo, el trastorno representado en el árbol genealógico
de la fi gura 7-8 posiblemente sea un rasgo autosómico recesi-
vo, a pesar de que el resto de la información relativa al árbol
genealógico puede parecer insufi ciente para establecer este pa-
trón de herencia.
El riesgo genético que presentan los descendientes de
progenitores con consanguinidad no es tan elevado como el
que se supone en ocasiones. En lo que se refi ere a los matri-
monios entre primos hermanos, los riesgos absolutos de que
los descendientes tengan problemas (considerando no sólo las
enfermedades autosómicas recesivas conocidas, sino también
los abortos, los cuadros de fallecimiento en la fase neonatal y
las malformaciones congénitas) es del 3-5%, es decir, aproxi-
madamente el doble del riesgo global básico del 2-3% que pre-
sentan los hijos de parejas que no muestran consanguinidad
(v. cap. 19). La consanguinidad a nivel de primos terceros o de
familiares más remotos no se considera genéticamente signifi -
cativa y en estos casos el aumento en el riesgo de alteraciones
en la descendencia es despreciable.
Aunque la incidencia del matrimonio entre primos es hoy
día baja (aproximadamente, el 1-10 por 1.000) en muchos gru-
pos de población de los países occidentales esta práctica sigue
siendo relativamente frecuente en algunos grupos raciales; por
ejemplo, en las familias de las áreas rurales del subcontinente
hindú, en otras partes de Asia y en Oriente Medio, en donde
el 20-60% de todos los matrimonios tiene lugar entre primos.
No obstante, en términos generales, la frecuencia de los matri-
monios entre primos hermanos y de la consanguinidad en ge-
neral está disminuyendo en muchas sociedades tradicionales.
La consanguinidad no es la explicación más habitual de
un rasgo autosómico recesivo. El emparejamiento de personas
genéticamente no relacionadas que son portadoras por azar
explica la mayor parte de los casos de enfermedades autosó-
micas recesivas, especialmente si el rasgo recesivo presenta una
frecuencia elevada en la población. Así, en la mayor parte de
las personas afectadas que sufren un trastorno relativamen-
te frecuente, tal como la CF, el problema no es el resultado
de la consanguinidad, debido a que el alelo mutante muestra
una frecuencia elevada en la población general. Sin embargo,
la consanguinidad es más frecuente como causa de fondo en
los pacientes que sufren enfermedades muy infrecuentes. Por
ejemplo, en la xerodermia pigmentosa
Caso 43) (un trastor-
no autosómico recesivo muy infrecuente de la reparación del
DNA; v. cap. 16), más del 20% de los casos tiene lugar entre la
descendencia de matrimonios entre primos hermanos.
Determinación de la consanguinidad
La determinación de la consanguinidad es relevante en genéti-
ca médica debido a que el riesgo de que un niño sea homoci-
goto respecto a un alelo recesivo infrecuente es proporcional al
grado de relación genética existente entre sus padres. Algunos
tipos de matrimonios consanguíneos conllevan un aumento
del riesgo (fi g. 7-9).
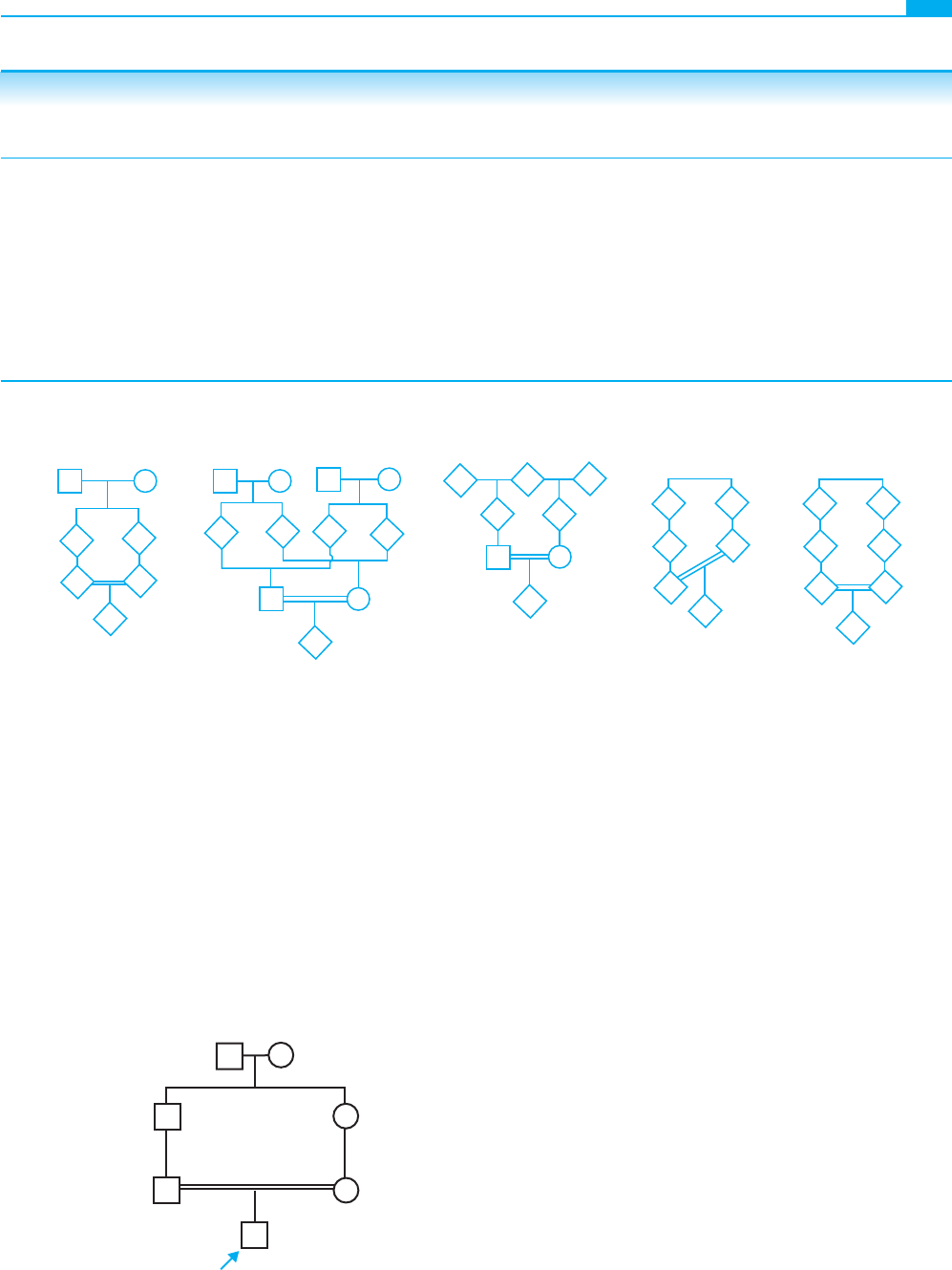
La consanguinidad se determina mediante el coefi ciente
de endogamia (F), que indica la probabilidad de que un homo-
cigoto haya recibido los dos alelos correspondientes a un locus
a través de un mismo antepasado; también es la proporción
de loci respecto a los cuales una persona es homocigota para
un alelo procedente del mismo antepasado, una situación de-
nominada identidad por ascendencia. En la fi gura 7-10, el in-
dividuo IV-1 es hijo de una pareja de primos hermanos. Cada
uno de los cuatro alelos del locus A (A
1
, A
2
, A
3
y A
4
) en la
generación I tiene una probabilidad 1/8 × 1/8 = 1/64 de ser
homocigoto en IV-1; por tanto, la probabilidad de que IV-1
sea homocigoto para cualquiera de los cuatro alelos es 4 × 1/64 =
1/16. La tabla 7-1 muestra los coefi cientes de endogamia de
los descendientes de una serie de emparejamientos consanguí-
I
II
III
IV
Figura 7-8 ■ Árbol familiar en el que la consanguinidad
de los padres sugiere una herencia autosómica recesiva.
CAPÍTULO 7
●
Patrones de herencia monogénica
125
© Elsevier. Es una publicación MASSON. Fotocopiar sin autorización es un delito.
neos. Si una persona presenta endogamia a través de más de
una línea de ascendencia, se suman los coefi cientes separados
para calcular su coefi ciente total de endogamia. (v. el problema
7 al fi nal de este capítulo.)
El consejo genético relativo al riesgo de malformaciones
congénitas y de enfermedades genéticas en los hijos de parejas
consanguíneas se expone en el capítulo 19.
Endogamia
La endogamia está estrechamente relacionada con la consan-
guinidad. Este concepto describe la situación en la que los in-
dividuos pertenecientes a una población de tamaño pequeño
tienden a seleccionar a sus parejas en la misma población,
debido a razones culturales, geográfi cas o religiosas. En esta
situación, los padres podrían no estar genéticamente relacio-
nados y, a pesar de ello, tener un antecesor común en las ge-
neraciones anteriores. De la misma manera que ocurre con la
consanguinidad, la endogamia incrementa las posibilidades de
que los individuos sean homocigotos para un alelo heredado a
partir de un antecesor común. Así, cuando se realiza la historia
clínica, es importante preguntar no solamente por la consan-
guinidad sino también por los orígenes geográfi cos de los ante-
cesores, especialmente si la pareja que solicita consejo procede
de un contexto racial o geográfi co similar. De la misma forma
que ocurre con los emparejamientos consanguíneos, es posible
calcular un coefi ciente de endogamia en las personas de una
población, a pesar de que ellas mismas no sepan que están
genéticamente relacionadas entre sí.
Aunque por lo general se establece la distinción entre la
consanguinidad de una familia y su nivel de endogamia, que
tiene lugar entre los individuos genéticamente no relacionados
Primos segundos
Primos hermanos
Primos hermanos dobles
Medio primos hermanos
Entre primos hermanos
y segundos
(
first cousins once removed
)
Figura 7-9 ■ Tipos de emparejamientos consanguíneos. La probabilidad de que los descendientes de cada uno de estos
emparejamientos sean homocigotos por su ascendencia en cualquier locus es igual al coefi ciente de endogamia, F.
I
II
III
IV
1
1
2
2
1
2
1
A
1
/A
2
A
3
/A
4
Figura 7-10 ■ Un matrimonio entre primos, utilizado en
el texto como ejemplo para calcular el coefi ciente de endoga-
mia, F, del niño IV-1.
Tabla 7-1
Emparejamientos consanguíneos
Proporción de genes Coefi ciente de endogamia
Tipo Grado de relación en común del niño (F)
Gemelos monocigóticos NA 1 NA
Progenitor-hijo Primer grado 1/2 1/4
Hermano-hermana (incluyendo gemelos dicigóticos) Segundo grado 1/2 1/4
Hermano-hermanastra Segundo grado 1/4 1/8
Tío-sobrina o tía-sobrino Segundo grado 1/4 1/8
Medio tío-sobrina Tercer grado 1/8 1/16
Primos hermanos Tercer grado 1/8 1/16
Primos hermanos dobles Segundo grado 1/4 1/8
Medio primos hermanos Cuarto grado 1/16 1/32
Entre primos hermanos y segundos Cuarto grado 1/16 1/32
Primos segundos Quinto grado 1/32 1/64
Coefi ciente de endogamia para la descendencia de diversos emparejamientos consanguíneos. Si una persona presenta endogamia través de más de una
línea de ascendencia, se deben sumar los distintos coefi cientes para calcular su coefi ciente total de endogamia. NA, no aplicable.
Thompson & Thompson GENÉTICA EN MEDICINA
126
pero pertenecientes a un mismo grupo racial de tamaño pe-
queño, en ambas situaciones existe un aumento en el riesgo de
emparejamientos entre portadores heterocigotos de trastornos
autosómicos recesivos.
Trastornos recesivos infrecuentes en grupos
genéticamente aislados
Existen numerosos grupos de tamaño pequeño en los que la
frecuencia de algunos genes recesivos infrecuentes es superior
a la que se observa en la población general. Estos grupos, de-
nominados grupos con aislamiento genético, pueden haber
quedado separados de los grupos vecinos debido a barreras
geográfi cas, religiosas o lingüísticas. A pesar de que estas po-
blaciones no son consanguíneas, las posibilidades de empare-
jamientos con otro portador de un trastorno recesivo concreto
pueden ser tan elevadas como las que se observan entre primos
hermanos.
La enfermedad de Tay-Sachs (gangliosidosis GM
2
) es un
ejemplo de enfermedad autosómica recesiva con un aumen-
to de su frecuencia en ciertos grupos genéticamente aislados
(Caso 38)
. Es un trastorno neurológico degenerativo que se
inicia cuando el niño tiene aproximadamente 6 meses de edad.
Los niños afectados sufren ceguera asociada a retraso mental y
físico (v. cap. 12), y la enfermedad es mortal durante la prime-
ra niñez. Por ejemplo, entre los judíos asquenazíes de América
del Norte la enfermedad de Tay-Sachs es 100 veces más fre-
cuente (1/3.600) que en otros grupos de ascendencia europea.
Este incremento en la incidencia de la enfermedad se debe a
que la frecuencia de portadores de la misma entre los judíos
asquenazíes (aproximadamente, 1/30) es 10 veces mayor que
en poblaciones europeas similares no constituidas por judíos
de esta etnia (el cálculo de ello se describe en el cap. 9).
Cuando los alelos mutantes que causan una enfermedad
recesiva son relativamente frecuentes en un grupo de población
concreto, los cónyuges genéticamente no relacionados mues-
tran una probabilidad razonable de ser heterocigotos y, por
tanto, la consanguinidad no es algo infrecuente en las familias
con niños afectados. Por ejemplo, entre los judíos asquenazíes,
los padres de niños con enfermedad de Tay-Sachs no suelen
mantener entre sí una relación genética muy estrecha. Sin em-
bargo, cuando el alelo mutante es infrecuente, la frecuencia de
los portadores es muy baja y a menudo es la consanguinidad la
explicación de que ambos miembros de una pareja sean hete-
rocigotos. Por ejemplo, se observa frecuentemente consangui-
nidad en los padres de pacientes con enfermedad de Tay-Sachs
pertenecientes a las poblaciones de ascendencia francesa de
Quebec, Canadá, en las que los alelos mutantes para la enfer-
medad de Tay-Sachs son infrecuentes.
Mutaciones nuevas en las enfermedades autosómicas
recesivas
Cuando un niño presenta afectación por una enfermedad au-
tosómica recesiva, se suele asumir que sus padres son porta-
dores heterocigotos para la misma (v. recuadro). No obstante,
las mutaciones nuevas se producen en todo momento durante
la generación de los gametos (v. cap. 9). ¿Sería posible que un
individuo presentara dos alelos mutantes para una enferme-
dad autosómica recesiva debido a que ha heredado un alelo
mutante a partir de uno de sus progenitores que es portador,
mientras que el otro alelo mutante se ha originado de novo en
un gameto procedente de un progenitor que no era portador?
Por supuesto, esta situación no es imposible pero es relativa-
mente improbable en comparación con la situación en la que
ambos progenitores son portadores heterocigotos. La razón
de ello es que la probabilidad de que el gameto procedente de
un progenitor no portador haya adquirido un alelo mutante a
través de una mutación espontánea oscila entre 1/10
5
y 1/10
6
(v. cap. 9), es decir, miles de veces menor que la probabilidad
típica de 1/20-1/1.000 de que el gameto contenga el alelo mu-
tante debido a que el progenitor es un portador heterocigoto.
La relativa falta de importancia de las mutaciones nuevas en lo
que se refi ere a las enfermedades autosómicas recesivas es una
situación completamente distinta a la que tiene lugar respecto
a los trastornos dominantes y ligados al cromosoma X, que
serán expuestos más adelante en este capítulo.
Herencia autosómica dominante
Más de la mitad de todos los trastornos mendelianos se here-
da en forma de rasgos autosómicos dominantes. La incidencia
de algunos trastornos autosómicos dominantes es elevada, al
menos en algunas áreas geográfi cas específi cas; por ejemplo,
es de 1/500 para la hipercolesterolemia familiar
(Caso 14) en
grupos de ascendencia europea o japonesa; de 1/550 para la
distrofi a miotónica en las regiones Charlevoix y Saguenay-
Lac Saint Jean del noreste de Quebec, y de aproximadamente
1/2.500-3.000 para trastornos graves como la enfermedad de
Huntington
(Caso 22) en poblaciones de ascendencia nórdi-
ca europea, para la neurofi bromatosis (Caso 29) y para la
enfermedad renal poliquística
(Caso 32) . A pesar de que mu-
chos trastornos autosómicos dominantes son individualmente
muy poco frecuentes, son tan numerosos en conjunto que su
incidencia total es apreciable. La incidencia de enfermedades
autosómicas dominantes es todavía mayor a consecuencia de
su carácter hereditario; cuando se transmiten a través de las
familias, se convierten en un problema no solamente para los
individuos afectados sino también para todo el árbol familiar,
a menudo a lo largo de múltiples generaciones. En algunos
casos, el problema representado por estas enfermedades se
● ■ ●
Características de la herencia autosómica
recesiva
• Un fenotipo autosómico recesivo, cuando aparece en
más de un miembro de un grupo familiar, se observa
característicamente sólo en los hermanos del probando,
no en los padres, los hijos ni otros familiares.
• En lo que se refi ere a la mayor parte de las enferme-
dades autosómicas recesivas, los individuos de ambos
sexos tienen una probabilidad similar de presentar
afectación.
• Los padres de un niño afectado son portadores asin-
tomáticos de alelos mutantes.
• Los padres de la persona afectada pueden ser en algunos
casos consanguíneos. Esta situación es especialmente
probable y el gen responsable del trastorno es infre-
cuente en la población general.
• El riesgo de recurrencia en cada hermano del probando
es de 1/4.

CAPÍTULO 7
●
Patrones de herencia monogénica
127
© Elsevier. Es una publicación MASSON. Fotocopiar sin autorización es un delito.
complica por las difi cultades sociales que acompañan a la dis-
capacidad física o mental.
El riesgo y la gravedad de las enfermedades de herencia
autosómica en la descendencia dependen de que estén afecta-
dos uno o los dos progenitores y de que el rasgo transmitido
sea dominante puro o dominante incompleto. Si denominamos
D al alelo mutante y d al alelo normal, los emparejamientos
que dan como resultado hijos que sufren la enfermedad auto-
sómica dominante pueden tener lugar entre dos heterocigotos
(D/d) para la mutación o bien, lo más frecuente, entre un he-
terocigoto para la mutación (D/d) y un homocigoto para un
alelo normal (d/d):
Riesgo para la
Emparejamientos Descendencia descendencia
Afectado con no afectado, 1/2 D/d, 1/2 afectado
D/d × d/d 1/2 d/d 1/2 no afectado
Afectado con afectado, 1/4 D/D, Si es dominante puro:
D/d × D/d 1/2 D/d, 3/4 afectados
1/4 d/d 1/4 no afectado
Si es dominante
incompleto:
1/2 afectado con la
misma intensidad
que los padres
1/4 afectado con una
intensidad mayor
que los padres
1/4 no afectado
Cada hijo de un emparejamiento D/d x d/d presenta una
probabilidad del 50% de recibir el alelo D anómalo de sus pa-
dres y una posibilidad del 50% de recibir el alelo d normal. Si
tomamos la población en conjunto, la descendencia de los em-
parejamientos D/d x d/d es D/d en aproximadamente el 50%
de los casos y d/d en alrededor del 50% restante. Por supues-
to, cada embarazo es un acontecimiento independiente que no
tiene relación con el resultado de los embarazos previos. Por
tanto, la distribución de los hijos afectados y no afectados en
una familia puede ser muy diferente del cociente teórico espe-
rado de 1:1, especialmente si el número de descendientes es pe-
queño. Se puede observar una herencia autosómica dominante
típica en el árbol genealógico de una familia con la forma here-
ditaria dominante de la sordera congénita (fi g. 7-11A).
En la práctica médica, los homocigotos para los fenotipos
dominantes no son frecuentes debido a que los emparejamien-
tos que podrían dar lugar a hijo homocigoto son infrecuentes.
De nuevo, si consideramos que D es el alelo mutante y d el
alelo normal, los emparejamientos que pueden dar lugar a un
homocigoto D/D podrían ser teóricamente D/d x D/d, D/D x
D/d y D/D x D/D; además, en situaciones extraordinariamente
infrecuentes, el paciente podría haber recibido una mutación
nueva por parte de un progenitor genéticamente no afectado.
Sin embargo, en términos prácticos, sólo se deben considerar
los emparejamientos entre dos heterocigotos debido a que los
homocigotos D/D son muy infrecuentes y a que generalmente
están demasiado afectados como para reproducirse (capaci-
dad reproductiva = 0). En el caso de un emparejamiento entre
dos heterocigotos, 3/4 hijos de emparejamientos D/d x D/d
van a presentar afectación en alguna medida, y 1/4 no va a
presentar afectación. Teóricamente, los 3/4 afectados podrían
presentar el mismo tipo de enfermedad si su transmisión fuera
dominante pura mientras que el 1/3 de los afectados podría
ser homocigoto y mostrar una afectación mucho más intensa
que la de los heterocigotos D/d, en el caso de que la enferme-
dad fuera un trastorno dominante incompleto. De hecho, tal
como ya se ha mencionado, no se ha demostrado que ninguno
de los trastornos dominantes que afectan al ser humano sea
dominante puro. Incluso la enfermedad de Huntington, que
es el trastorno del que se suele decir con mayor frecuencia que
es dominante puro debido a que generalmente la enfermedad
muestra unas características y una sintomatología de intensi-
dad similares en los heterocigotos y los homocigotos, parece
presentar en los homocigotos una cierta evolución cronológica
acelerada desde el comienzo del proceso hasta el fallecimiento,
en comparación con los heterocigotos.
Herencia dominante incompleta
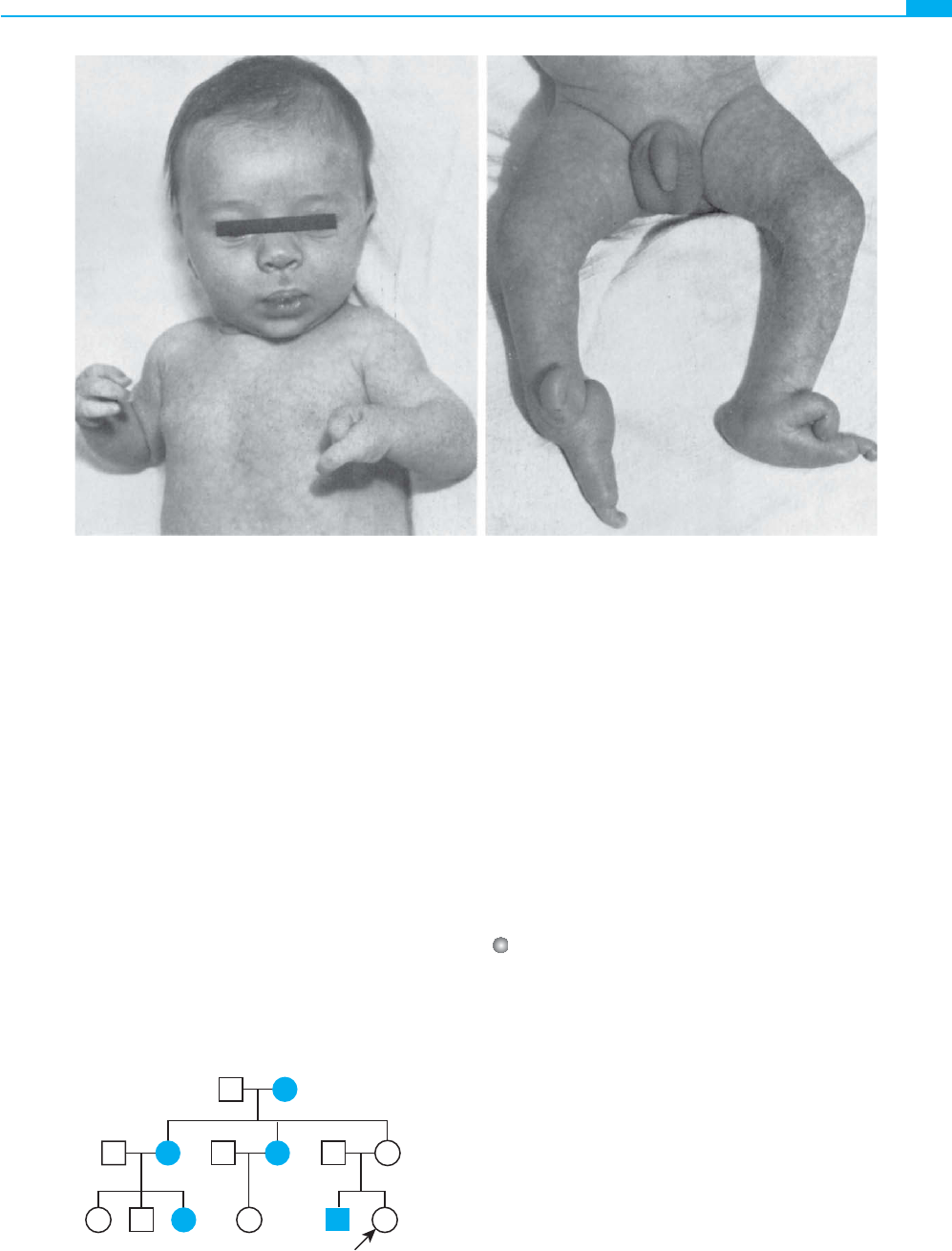
La acondroplasia es un trastorno esquelético dominante incom-
pleto que causa un enanismo con miembros cortos y con cabe-
za grande (fi g. 7-12 )
(Caso 1) . La mayor parte de los pacien-
tes con acondroplasia presenta una inteligencia normal y lleva
a cabo una vida también normal dentro de sus limitaciones fí-
sicas. Los matrimonios entre dos pacientes con acondroplasia
no son infrecuentes. El hijo homocigoto de dos heterocigotos
se puede reconocer generalmente a través de las manifestacio-
nes clínicas; los individuos homocigotos para la acondroplasia
I
II
III
IV
A
I
II
III
B
2
3
Figura 7-11 ■ A: Árbol familiar en el que se observa la herencia típica de una forma de sordera neurosensitiva progresiva
(DFNA1) heredada a través de un rasgo autosómico dominante. B: Árbol genealógico en el que se observa la herencia de la
acondroplasia, un rasgo dominante incompleto (o semidominante).
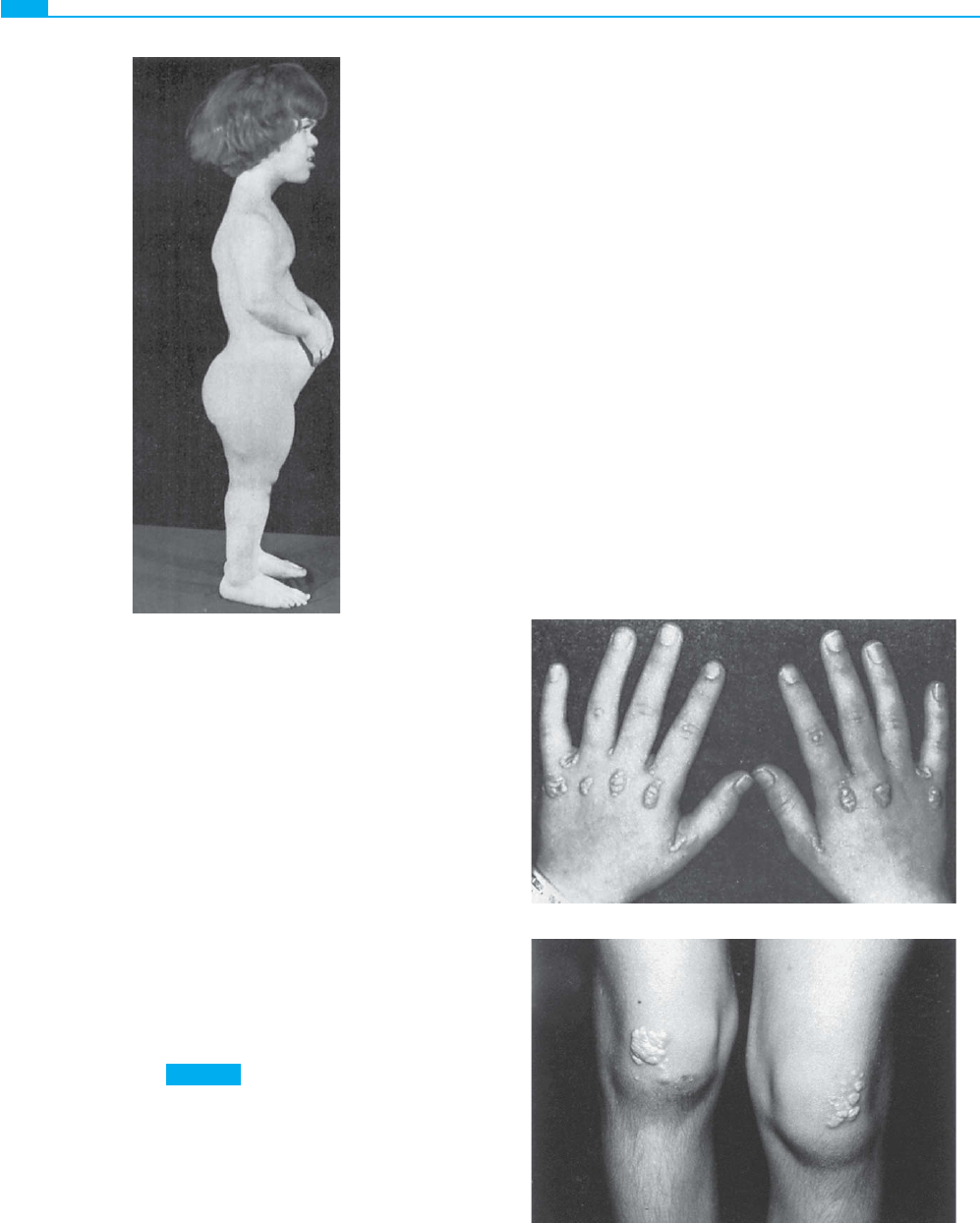
Thompson & Thompson GENÉTICA EN MEDICINA
128
muestran una afectación mucho más intensa que los heteroci-
gotos y generalmente no sobreviven más allá del periodo pos-
natal inmediato. En la fi gura 7-11B se muestra el árbol fami-
liar correspondiente al emparejamiento entre dos individuos
heterocigotos para la mutación que causa la acondroplasia. El
niño fallecido (el individuo III-3) era un homocigoto para la
enfermedad y sufría un trastorno mucho más grave que cual-
quiera de sus progenitores, lo que dio lugar a su fallecimiento
al poco de nacer.
Otro ejemplo de trastorno dominante incompleto es la
hipercolesterolemia familiar (v. cap. 12), una enfermedad au-
tosómica dominante que da lugar a la aparición de una coro-
nariopatía prematura
(Caso 14) . En este trastorno, los escasos
pacientes homocigotos sufren un cuadro de mayor gravedad en
el que la enfermedad se inicia a una edad más temprana (con
reducción de la esperanza de vida), en comparación con los
heterocigotos que son relativamente más comunes (fi g. 7-13).
Mutaciones nuevas en la herencia autosómica
dominante
En la herencia autosómica dominante típica, todas las perso-
nas afectadas de un árbol familiar tienen un progenitor afec-
tado que, a su vez, también tiene un progenitor afectado y así
sucesivamente de forma retrospectiva hasta donde es posible
seguir la enfermedad o hasta que se demuestra la aparición
de una mutación original. Tal como se expone más adelante,
esta secuencia también es cierta en los árboles genealógicos de
enfermedades dominantes ligadas al cromosoma X. De hecho,
la mayor parte de los trastornos dominantes con una cierta im-
portancia médica tiene lugar no solamente a través de la trans-
misión del alelo mutante por parte de un progenitor portador,
sino también a través de la herencia de una mutación nueva y
espontánea en un gameto heredado a partir de un progenitor
que no es heterocigoto. La razón es que los trastornos domi-
nantes pueden manifestarse en los casos en los que sólo está
alterado uno de los componentes del par de alelos, tanto si este
alelo se hereda a partir de un progenitor heterocigoto como
si procede de una mutación nueva y espontánea en el gameto
transmitido por un progenitor no heterocigoto (v. fi g. 7-11B).
Relación entre las mutaciones nuevas y la capacidad
reproductiva en los trastornos autosómicos
dominantes
Una vez que se ha producido una mutación nueva, su supervi-
vencia en la población depende de la capacidad reproductiva de
las personas portadoras de la misma, es decir, de la capacidad
de un heterocigoto para reproducirse y transmitir así el nuevo
alelo mutante. Hay una relación inversa entre la capacidad
reproductiva de un trastorno autosómico dominante dado y
Figura 7-12 ■ Acondroplasia, un trastorno autosómico
dominante que aparece con frecuencia debido a una mutación
nueva. Se pueden observar la estatura corta, los miembros
cortos, la cabeza grande, el puente nasal hundido, la frente
prominente y la lordosis lumbar, en este caso típico. (Tomada
de Tachdjian MO: Pediatric Orthopedics, vol. 1. Filadelfi a,
WB Saunders, 1972, pág. 284.)
A
B
Figura 7-13 ■ Xantomas cutáneos en un homocigoto para
la hipercolesterolemia familiar. (A: cortesía de J. L. Goldstein,
University of Texas Southwestern Medical Center, Dallas. B:
tomada de Teruel JL, Lasunción MA: Cutaneous xanthomas
in homozygous familial hypercholesterolemia. N Engl J Med
332:1137, 1995. © Reservados todos los derechos.)
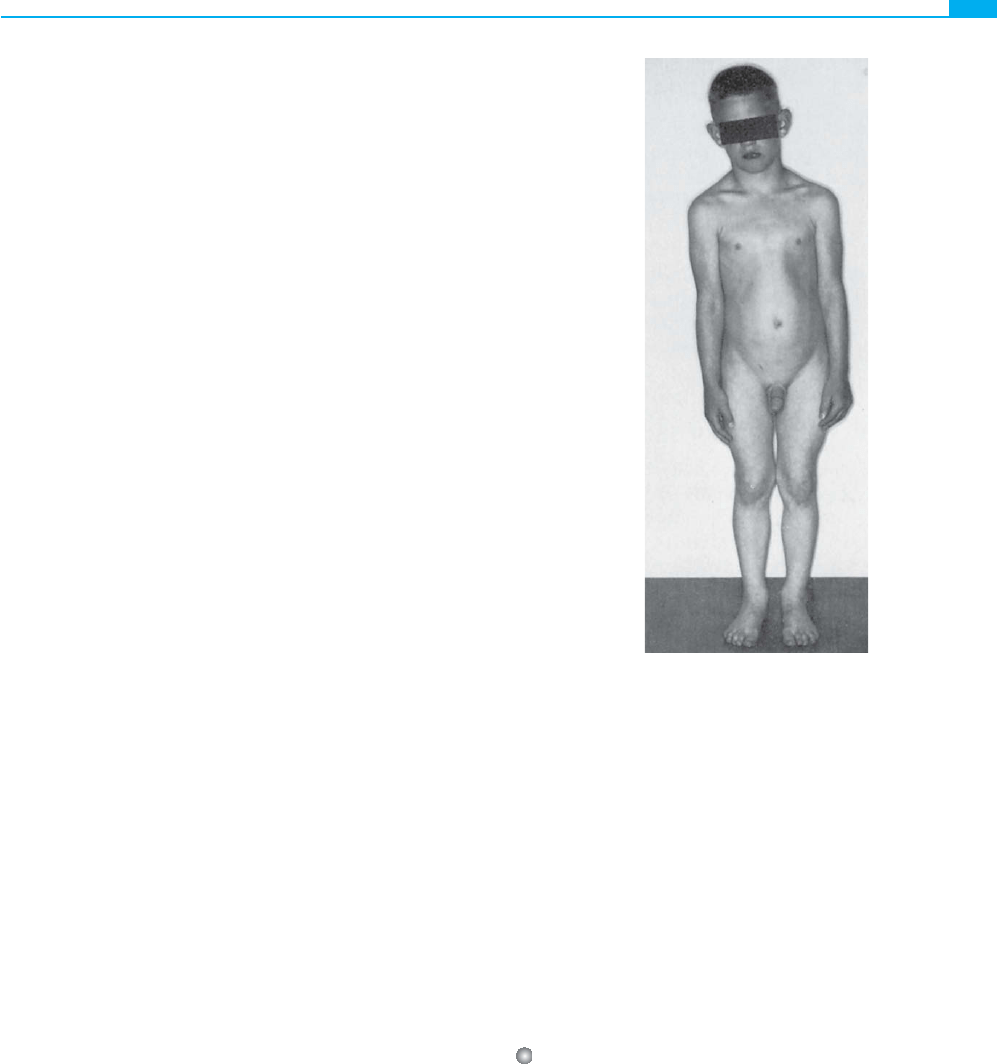
CAPÍTULO 7
●
Patrones de herencia monogénica
129
© Elsevier. Es una publicación MASSON. Fotocopiar sin autorización es un delito.
la proporción de todos los pacientes que presentan la enferme-
dad y que recibieron el gen alterado en forma de una mutación
nueva, más que en forma de la transmisión procedente de un
progenitor heterocigoto. En un extremo se sitúan los trastor-
nos con una capacidad reproductiva de 0; en otras palabras,
los pacientes que sufren estos problemas no se reproducen y
se considera que, por tanto, el trastorno es genéticamente le-
tal. Todas las enfermedades autosómicas dominantes genéti-
camente letales tienen que ser debidas a mutaciones nuevas
ya que estas mutaciones no se transmiten hereditariamente.
Los individuos afectados aparecen como casos aislados en el
árbol genealógico. En el otro extremo están las enfermedades
con una capacidad reproductiva virtualmente normal debido a
que se inician en etapas tardías de la vida o a que cursan con
un fenotipo leve que no interfi ere con la reproducción. Si la
capacidad reproductiva es normal, la enfermedad no suele ser
el resultado de una mutación reciente; en estos casos es mucho
más probable que el paciente haya heredado la enfermedad
en comparación con la posibilidad de que haya presentado
una mutación nueva, y el árbol genealógico posiblemente va
a incluir a múltiples individuos afectados con un patrón de
herencia autosómica dominante claro. La determinación de la
frecuencia de la mutación y de la relación existente entre la fre-
cuencia de la mutación y la capacidad reproductiva se expone
con mayor detalle en el capítulo 9.
Fenotipo con limitación sexual en las enfermedades
autosómicas dominantes
Tal como se ha señalado antes en relación con la hemocro-
matosis autosómica recesiva, los fenotipos autosómicos do-
minantes también pueden presentar variaciones sexuales que
se apartan signifi cativamente de la proporción 1:1. Hay una
divergencia extrema en la proporción de sexos en los fenoti-
pos con limitación sexual, en los que el defecto se transmite
de manera autosómica pero sólo es expresado por uno de los
dos sexos. Un ejemplo de ello es la pubertad precoz limitada a
los individuos de sexo masculino (testotoxicosis familiar), un
trastorno autosómico dominante en el que los niños afectados
desarrollan los caracteres sexuales secundarios y presentan el
estirón de crecimiento de la adolescencia aproximadamente a
los 4 años de edad (fi g. 7-14). En algunas familias, este defecto
se ha puesto en relación con mutaciones en el gen que codifi ca
el receptor de la hormona luteinizante (LCGR); estas muta-
ciones activan de manera constitutiva el mecanismo de señales
del receptor incluso en ausencia de su hormona. El defecto no
se manifi esta en las mujeres heterocigotas. En el árbol fami-
liar que se muestra en la fi gura 7-15 se puede observar que,
aunque la enfermedad puede ser transmitida por mujeres no
afectadas, también se puede transmitir directamente desde un
padre a su hijo de sexo masculino, lo que demuestra que es
autosómica y no ligada al cromosoma X.
Los niños de sexo masculino con pubertad precoz debido
a la activación de mutaciones LCGR presentan una fertilidad
normal y se han descrito numerosos árboles genealógicos mul-
tigeneracionales de este tipo. No obstante, en lo que se refi ere
a los trastornos en los que los individuos de sexo masculino
afectados no pueden reproducirse, no siempre es fácil diferen-
ciar la herencia autosómica con limitación sexual de la heren-
cia ligada al cromosoma X, dado que no se puede demostrar
el dato de carácter crítico (la ausencia de transmisión entre
individuos de sexo masculino). En este caso, otras líneas de
evidencia (especialmente, la cartografía de genes para deter-
minar si el gen responsable se localiza en el cromosoma X o
en un autosoma [v. cap. 10]) permiten determinar el patrón de
herencia y el riesgo consiguiente de recurrencia (v. recuadro,
pág. 130).
HERENCIA LIGADA AL CROMOSOMA X
Los cromosomas X e Y, que son los responsables de la deter-
minación sexual (v. cap. 6), se distribuyen de manera desigual
a los hombres y a las mujeres en las familias. Por esta razón,
los fenotipos determinados por los genes localizados en el cro-
mosoma X muestran una distribución sexual y un patrón de
herencia característicos que generalmente permiten su identifi -
cación con facilidad. Se considera que hay aproximadamente
1.100 genes localizados en el cromosoma X, de los cuales sa-
bemos en la actualidad que alrededor del el 40% se asocia a
fenotipos de enfermedad.
Dado que los individuos de sexo masculino sólo poseen un
cromosoma X, mientras que los de sexo femenino poseen dos,
sólo hay dos posibles fenotipos en los hombres y tres posibles
fenotipos en las mujeres, en lo que se refi ere a un alelo mutante
Figura 7-14 ■ Pubertad precoz limitada al sexo masculino
(testotoxicosis familiar), un trastorno autosómico dominante
expresado únicamente por los individuos de sexo masculino.
Este niño de 4,75 años de edad tiene una estatura de 120 cm
(por encima del percentil 97 para su edad). Se pueden observar
los músculos voluminosos y el desarrollo precoz de los geni-
tales externos. La fusión de las epífi sis tiene lugar a una edad
temprana y las personas afectadas muestran una estatura rela-
tivamente corta cuando alcanzan la edad adulta.
Este documento contiene más páginas...
Descargar Completo
Capitulo7-Patrones de herencia monogenica_2008_ThompsonThompson (1).pdf
 Estamos procesando este archivo...
Estamos procesando este archivo...
 Lamentablemente la previsualización de este archivo no está disponible. De todas maneras puedes descargarlo y ver si te es útil.
Lamentablemente la previsualización de este archivo no está disponible. De todas maneras puedes descargarlo y ver si te es útil.
Descargar
Estamos procesando este archivo...
Lamentablemente la previsualización de este archivo no está disponible. De todas maneras puedes descargarlo y ver si te es útil.