
Trastornos
hemodinámicos.
Edemas y derrames.
Edema: acumulación de líquido en los tejidos.
Derrame: acumulación de líquido en cavidades corporales.
Habitualmente hay un pequeño movimiento de liquido al intersticio, pero se drena a los linfáticos y vuelve
al torrente sanguíneo por el conducto torácico. El aumento de presión hidrostática o la reducción de la
presión coloidosmotica alteran este equilibrio y permiten una mayor salida de liquido de los vasos. Si la
tasa de salida de liquido supera la tasa de drenaje linfático, el liquido se acumula.
EDEMAS Y DERRAMES INFLAMATORIOS
EDEMAS Y DERRAMES NO INFLAMATORIOS
Exudados ricos en proteínas
Trasudados con escasas proteínas
Aumento de la permeabilidad por mediadores
inflamatorios
Otras causas como alteración de las fuerzas de
Starling
Pueden ser locales o generalizados en la sepsis.
Se ven en insuficiencia cardiaca, insuficiencia
hepática, nefropatías y trastornos nutricionales
graves
CAUSAS DE EDEMAS Y DERRAMES.
1. Aumento de la presión hidrostática. Causado frecuentemente por alteración del retorno venoso, si
es localizada el edema queda limitado a esa parte, si las enfermedades provocan un aumento
sistémico de la presión venosa el edema es diseminado.
2. Reducción de la presión osmótica del plasma. En condiciones normales esa presión esta dada
principalmente por la albumina, la proteína plasmática mas abundante. Las situaciones que
disminuyan la albumina por perdida o menor síntesis generan disminución de la presión osmótica.
La síntesis se ve afectada en enfermedades hepáticas graves. La perdida se puede dar por el
síndrome nefrítico. Provoca edemas, menor volumen intravascular, hipoperfusion renal e
hiperaldosteronismo.
3. Retención de sodio y agua. El aumento de retención de sal, con su retención forzosa de agua
asociada, causa un aumento de la Ph y disminución de la presión coloidosmotica vascular. La
retención de sal se produce en situaciones con función de sal alterada y trastornos
cardiovasculares que reduzcan la perfusión renal.
4. Obstruccion linfática. Por traumatismos, fibrosis, tumores infiltrantes y algunos microorganismos
infecciosos, se altera el proceso de eliminación del liquido intersticial, dando lugar a linfedema en la
parte afectada del cuerpo. Esto se observa en la elefantiasis (inflamación de genitales y MMII por
fibrosis obstructiva de vasos y ganglios linfáticos).
MORFOLOGIA DEL EDEMA.
Microscópicamente es una aclaración con separación de la matriz extracelular y leve tumefacción
celular. Aparece con frecuencia en tejidos subcutáneos, pulmones y encéfalo.
Subcutaneo: difuso o localizado, su distribución se ve influida por la gravedad, característica
denominada edema ortostatico (aparece en las piernas al estar de pie y el sacro al acostarse).
Edema con fóvea: cuando la presión del dedo sobre un tejido subcutáneo muy edematoso
desplaza el liquido intersticial y deja una cavidad.
Secundario a disfunción renal: suele aparecer en partes con TCL como parpados (edema
periorbitario, característico en nefropatía grave).

Edema de pulmón: los pulmones tienen dos o tres veces su eso y al corte aparece un liquido
espumoso y sanguinolento (mezcla de aire, edema y eritrocitos extravasados)
Edema cerebral: el encéfalo muestra estrechamiento de los surcos y distensión de las
circunvoluciones, que resultan comprimidas contra el cráneo rigido.
Derrames que afectan cavidades: Hidrotorax (derrame pleural), hidropericardio, hidroperitoneo
también llamado ascitis
Derrames trasudados pobres en proteinas, transparents y de color rojizo, excepto el peritoneal
causado por bloqueo linfático que puede ser lechoso.
Derrames exudados ricos en proteinas y turbios por la presencia de leucocitos.
CARACTERISTICAS CLÍNICAS.
El edema subcutáneo apunta a posibles enfermedades cardiacas o renales subyacentes, si es
intenso puede alterar la cicatrización de heridas o la erradicación de infecciones.
El edema de pulmón es observado en insuficiencia del Vent izq, insuficiencia renal, síndrome de
dificultad respiratoria aguda, e inflamaciones/ infecciones pulmonares.
Derrames pleurales: acompañan al de pulmón y comprometen aun mas el intercambio gaseoso.
Derrames peritoneales (ascitis): por hipertensión portal.
Edema cerebral: si es intenso el encéfalo se hernia a través del agujero occipital.
Hiperemia y congestión.
Ambos se producen por el aumento de volumen sanguíneo dentro de los tejidos, pero su mecanismo
subyacente es distinto, también sus consecuencias.
Como consecuencia del aumento de presión hidrostática la congestión suele seguirse de edema. En la
congestion crónica puede haber lesión tisular isquémica y cicatrices por hipoxia crónica, también rotura de
capilares que produce pequeños focos hemorrágicos; el catabolismo de esos eritrocitos extravasados deja
cúmulos de macrófagos cargados con hemosiderina.
MORFOLOGIA DE LA CONGESTION.
Los tejidos congestionados adoptan u color azul rojizo oscuro (cianosis) por la estasis de eritrocitos y
presencia de Hb desoxigenada.
Congestion pulmonar aguda: microscópicamente presenta capilares alveolares dilatados, edema
septal alveolar y hemorragias intraalveolares focales.
Congestion pulmonar crónica: causada en gral por ICC, se ve paredes engrosadas y fibroticas,
alveolos con numerosos macrófagos cargados de hemosiderina denominados celulas de la
insuficiencia cardiaca.
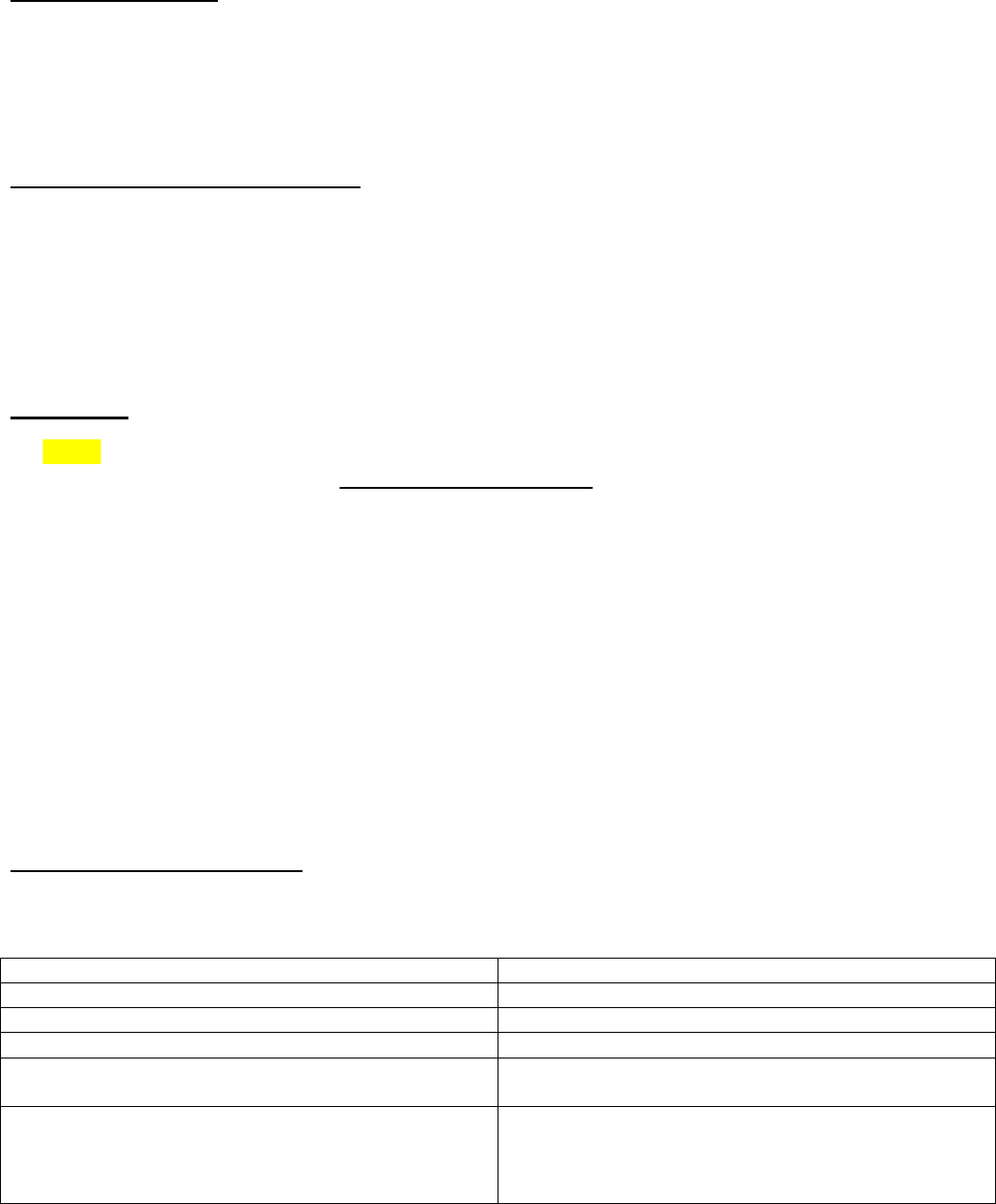
Congestion hepática aguda: vena central y sinusoides distendidos, los hepatocitos
centrolobulillares pueden sufrir necrosis isquémica, mientras que los periportales al estar mejor
oxigenados desarrollan degeneración grasa.
Congestion hepática pasiva crónica: macroscópicamente se ven regiones centrolobulillares de color
marron rojizo y ligeramente hundidas por muerte celular, y destacan en relación con zonas
circundantes de hígado marron no congestionado. A este patrón se lo llama higado en nuez
moscada. Microscopicamente hay hemorragia centrolobulillar, macrófagos con hemosiderina y
perdida y necrosis de hepatocitos.
Hiperemia es un proceso activo en el
que la dilatación arteriolar provoca
aumento del flujo saguineo, donde los
tejidos afectados se tornan rojos
(eritema) por el mayor aporte de
sangre.
Congestion es un proceso pasivo
secundario a una menor salida de
sangre de in tejido, puede ser
sistémica o localizada.

Hígado en nuez moscada.
Necrosis centrolobulillar y
hemorragia
Hemostasia, trastornos hemorrágicos y trombosis.
Hemostasia: proceso de formación de coágulos sanguíneos en zonas de lesión vascular.
Este proceso normal puede estar alterado en los trastornos hemorrágicos, donde hay sangrado excesivo
porque los mecanismos hemostáticos son insuficientes para prevenir perdidas de sangre, o puede estar
alterado en trastornos tromboticos, cuando se forman coágulos sanguíneos (trombos) en vasos
sanguíneos sin lesión alguna o en cavidades cardiacas.
Hemostasia.
Proceso en el que participan las plaquetas, factores de coagulación y endotelio. Tiene lugar en zonas de
lesión vascular y culmina con a formación de un coagulo, que previene o limita el alcance de la
hemorragia. Los procesos que suceden son:
1. Vasoconstriccion arteriolar.
2. Hemostsia primaria: formación del tapon plaquetario.
3. Hemostasia secundaria: deposito de fibrina.
4. Estabilizacion y reabsorción del coagulo.
1.Se produce una vasoconstricción inmediata que reduce mucho el flujo sanguíneo en el lugar de lesión,
es potenciada por la endotelina. 2. La rotura del endotelio expone el factor de von Willebrand (vWF) y
colágeno subendoteliales, promoviendo la adhesión y activación de plaquetas. La activación de plaquetas
hace que cambien de forma y liberen los granulos secretores. Los productos secretados reclutan aun mas
plaquetas que se agregan y forman el tapón hemostático primario. 3. Por otro lado, en la zona de lesión
también queda expuesto el factor tisular. El factor tisular es una glucoproteina procoagulante normalmente
expresada en celulas subendoteliales, del musculo liso y fibroblastos. Este factor se une al factor VII de la
cascada de la coagulación y lo activa, desencadenando la formación de trombina. La trombina transforma
el fibrinógeno en fibrina. Se crea una malla de fibrina y además, la fibrina activa las plaquetas, aumentando
la agregación plaquetaria. Asi se forma el tapon plaquetario inicial. 4. La fibrina y los agregados
plaquetarios se contraen para formar un tapon permanente solido que impide la hemorragia. En esa
ultima fase se activan mecanismos contrarreguladores que limitan la coagulación, provocan la reabsorción
del coagulo y reparación del tejido.
PLAQUETAS.
Son esenciales en la formación del tapon primario que sella inicialmente el defecto vascular, además de
brindar una superficie a la cual unirse a los factores de coagulación activados. Son fragmentos de celulas
sin nucleo en forma de disco que derivan de los megacariocitos de la medula osea y alcanzan la
circulación. Contienen dos tipos de granulos:
Granulos α: tienen en su interior fibrinógeno, factor V y vWF. Tambien fibronectina, factor
plaquetario 4, factor de crecimiento derivado de plaquetas y factor de crecimiento transformante β.
Granulos δ: contienen ADP y ATP, calcio, serotonina y adrenalina.
Tras la lesión vascular las plaquetas entran en contacto con vWF y colágeno, y pasan por las siguientes
reacciones.
Adhesion plaquetaria. Cambio rápido de forma. Secrecion.
Agregacion plaquetaria.
Adhesión plaquetaria: mediada por interacción con vWF, sirve de puente entre un receptor de
superficie de las plaquetas (glucoproteina Ib) y el colágeno expuesto.
Cambio de forma: las plaquetas pasan de ser discos lisos a forma de “erizo de mar” con un
incremento de la superficie. Este cambio altera la glucoproteina Ib, aumentando su afinidad con
fibrinógeno.
Secreción: se libera el contenido de los gránulos, se produce al mismo tiempo que el cambio de la
forma, ambos procesos, el cambio de forma y secreción implican la activación plaquetaria. La
activación plaquetaria esta desencadenada por distintos factores como trombina y ADP. Al ser el
ADP un componente de los gránulos, la liberación de los mismos genera nuevos ciclos de
activación plaquetaria, fenómeno denominado reclutamiento. Las plaquetas activadas producen
PG: el TxA2, potente inductor de la agregación plaquetaria.
Agregacion plaquetaria: luego de la activación. El cambio de la glucoproteina Ib permite la unión
con fibrinógeno. La activación concomitante de la trombina por exposición del factor tisular
estabiliza el tapón plaquetario al aumentar la activación y agregación y promover la contracción
plaquetaria irreversible. Al mismo tiempo la trombina convierte el fibrinógeno en fibrina,
cementando las plaquetas en su lugar y formando el tapon hemostático secundario definitivo.
CASCADA DE LA COAGULACIÓN.
Serie de reacciones enzimáticas amplificadoras que conduce a la formación de un coagulo de fibrina
insoluble. En cada reacción participan una enzima (factor de la coagulación activado), un sustrato
(proenzima inactiva de un factor de la coagulación) y un cofactor (acelerador de la reacción). Para la
coagulación se necesita Calcio y vitamina K. La cascada se divide en una via intrínseca y otra extrínseca.
Trombina. De los factores de la coagulación, la trombina es el mas importante, porque sus actividades
enzimáticas controlan diversos aspectos de la hemostasia y constituyen el nexo entre coagulación,
inflamación y reparación. Actividades mas importantes de la trombina.
Conversion de fibrinógeno en fibrina entrecruzada. Los monómeros de fibrina se polimerizan hasta
formar un coagulo insoluble y además amplifica el proceso de la coagulación. Tambien estabiliza el
tapon hemostático secundario.
Activacion plaquetaria. La trombina es un potente inductor de la activación y agregación
plaquetarias por su capacidad de activar los PAR (receptores presentes en las plaquetas).
Efectos proinflamatorios. Los PAR también se expresan en celulas inflamatorias, endotelio y otras,
y la trombina al unirse media efectos inflamatorios que contribuyen a la reparación tisular y
angiogenia.
Efectos anticoagulantes. La trombina pasa de ser procoagulante a anticoagulante al contactar con
un endotelio normal, lo que impide que el coagulo se extienda mas alla de la zona de lesión
vascular.
FACTORES QUE LIMITAN LA COAGULACIÓN.
La coagulación debe restringirse a la zona de lesion para impedir consecuencias perjudiciales. Factores
limitantes:
Dilucion: la sangre que fluye por la zona de lesión aleja los factores de coagulación activados, y
son eliminados por el hígado.
La activación de la cascada de coagulación también desencadena una cascada fibrinolitica que
limita el tamaño del coagulo y contribuye a su posterior disolución. La fibrinólisis es llevada a cabo
por plasmina, que degrada la fibrina e interviene en su polimerización.
ACTIVACION PLAQUETARIA.

ENDOTELIO.
El balance entre actividades anticoagulantes y procoagulantes del endotelio determina si se produce
formación, propagación o disolución del coagulo. Normalmente el endotelio expresa factores que inhiben
las acciones procoagulantes de las plaquetas y factores de coagulación, a la vez que promueven la
fibrinólisis. Tambien previenen la trombosis.
Por el otro lado, si el endotelio resulta dañado o expuesto a factores proinflamatorios las celulas
endoteliales pierden muchas propiedades antitromboticas.
Propiedades antitromboticas del endotelio normal:
Efectos inhibidores de las plaquetas: el endoteliosirve de barrera que impide el contacto de las
plaquetas con el vWF y el colágeno subendoteliales. Otros factores que libera el endotelio que
inhiben la activación y agregación plaquetarias son: PGI2, NO, adenosina difosfatasa. Tambien se
unen a la trombina y alteran su actividad.
Efectos anticuagulantes: el endotelio actua como barrera entre los factores de coagulación y el
factor tisular. Tambien expresa factores que se oponen a la coagulación: trombomodulina,
receptore de proteína C endotelial, moléculas similares a la heparina y el inhibidor de la via del
factor tisular
Efectos fibrinoliticos: las celulas endoteliales sintetizan t-PA, esencial en la via fibrinolitica.
Trastornos hemorrágicos.
Se deben a defectos primarios o secundarios en las paredes de los vasos, las plaquetas o los factores de
coagulación. Estan las hemorragias masivas asociadas a roturas de los grandes vasos.
Las enfermedades asociadas con hemorragias masivas y bruscas son: diseccion aortica en el síndrome de
Marfan, aneurisma de la aorta abdominal e infarto de miocardio complicado con rotura de la aorta o el
corazón.
Otros defectos leves de la coagulación solo se ponen de manifiesto en condiciones de sobrecarga
hemostatica como: parto, cirugía, intervenciones dentales, menstruación, traumatismos.
Causas frecuentes de tendencia hemorrágica: defectos del vWF, consumo de acetilsalicílico y uremia.
Deficits de factores de coagulación hereditarios.
Principios generales de las hemorragias:
Alteraciones de la hemostasia primaria (defectos en plaquetas o vWF): manifestadas por pequeñas
hemorragias en la piel o mucosas, en forma de petequias (1-2 mm) o purpura (>= 3mm). Las
hemorragias mucosas por alteraciones en la hemostasia primaria pueden gnerar epistaxis
(hemorragia nasal), hemorragia digestiva o menstruación excesiva (menorragia). También esta la
hemorragia intracerebral.
Alteraciones de la hemostasia secundaria (defectos en factores de coagulación): hemorragia de
partes blandas o articulaciones. Las hemorragias articulares (hemartros) tras traumas leves son
caracteristics de la hemofilia.
Alteraciones generalizadas con afectación de vasos pequeños: purpura palpable y equimosis. Las
equimosis son hemorragias de 1-2 cm de tamaño. En ellos la sangre extravasada es suficiente
para crear una masa palpable conocida como hematoma.
La relevancia clínica de la hemorragia depende del volumen, velocidad con la que se produce y su
localización. Perdidas mayores del 20% del volumen sanguíneo pueden causar shock hemorrágico. En el
encéfalo, como el cráneo es rigido, la hemorragia aumenta la presión intracraneal hasta comprometer la
irrigación sanguínea o provocar herniación del tronco del encéfalo. Las perdidas sanguíneas externas
seguidas pueden causar anemia ferropenica.
Trombosis.
Causada principalmente por:
Lesion endotelial
Estasis o flujo sanguíneo turbulento
Hipercoagulabilidad de la sangre
LESION ENDOTELIAL.
Causante de la activación plaquetaria, casi inevitablemente, luego de ella se forman trombos en el corazón
y circulación arterial, en los que la elevada velocidad del flujo sanguíneo impide la formación de coagulos
normalmente. Las lesiones endoteliales graves pueden desencadenar trombosis al exponer vWF y factor
tirular. Hay otros estimulos nocivos que promueven la trombosis al volver el endotelio protrombotico. Este
cambio se llama activación o disfunción endotelial, y puede producirse por lesiones físicas, infecciones,
flujo sanguíneo anómalo, mediadores de inflamación, alteraciones metabólicas y toxinas absorbidas del
humo de tabaco.
Alteraciones endoteliales protromboticas principales.
Cambios procoagulantes: las celulas endoteliales activadas por citosinas bajan la expresión de
trombomodulina (inhibidor de la coagulación). Esto permite una activación mantenida de la
trombina. Por otro lado, el endotelio activado regula a la baja otros anticoagulantes: proteína C e
inhibidor del factor tisular.
Efectos antifibrinoliticos. Las celulas endoteliales secretan inhibidores del activador de
plasminogeno. El plasminogeno activa la fibrinólisis, por lo tanto, esto no sucederá. Tambien se
regula a la baja la expresión de t-PA. Asi se desarrolla la formación de trombos.
ALTERACIONES DEL FLUJO SAGUINEO.
La turbulencia contribuye a trombosis arteriales y cardiacas causando lesión endotelial y formando
contracorrientes que contribuyen a zonas de estasis. La estasis contribuye al desarrollo de trombos
venosos. Normalmente el flujo es laminar, llevando las plaquetas por el centro del vaso, alejadas del
endotelio. La estasis y turbulencia provocan:
Activacion endotelial.
Alteracion del flujo laminar y puesta en contacto de plaquetas y endotelio.
Impiden la retirada de factores coagulantes.
El flujo sanguíneo alterado dispone a la trombosis por ejemplo en las placas ateroescleróticas,
aneurismas, infartos de miocardio, estenosis de la valvula mitral, hiperviscosidad (aumenta la resistencia al
flujo), anemia drepanocitica (los drepanocitos impiden el flujo sanguíneo en pequeños vasos generando
estasis).
HIPERCOAGULABILIDAD.
Tambien se la llama trombofilia, a cualquier trastorno de la sangre que predisponga a la trombosis. Es
importante en las trombosis venosas y se clasifica en trastornos primarios (genéticos) y secundarios
(adquiridos). Causas hereditarias: anomalías en el factor V y de la protrombina. Las personas con
predisposición genética pueden presentar trombosis en otras situaciones asociadas como gestación o
reposo en cama prolongado, inactividad forzosa como viajes largos en avión. Hay que tener en cuenta las
causas hereditarias de hipercoagulabilidad en pacientes menores de 50 años que tengan trombosis.
A diferencia de los trastornos hereditarios, la trombofilia adquirida suele ser multifactorial. Causas mas
importantes: estasis o lesión vascular. Hay hipercoagulabilidad secundaria a uso de anticonceptivos orales
o estado hiperestrogenico de la gestación. En el cáncer hay una predisposición por liberación de
procoagulantes por parte del tumor. Tambien la edad avanzada, tabaquismo y obesidad la promueven.
De los estados trombofilicos adquiridos, el síndrome de trombocitopenia inducida por heparina y el de
anticuerpos antifosfolipidicos son importantes problemas clínicos.
TRIADA DE VIRCHOW

Síndrome de trombocitopenia inducida por heparina. Aparece por administración de heparina no
fraccionada, que induce la aparición de anticuerpos que se unen a las plaquetas, condicionando su
activación, agregación y consumo (por eso se llama trombocitopenia). Asi se produce un estado
protrombotico.
Sindrome por anticuerpos antifosfolipidicos. Tiene manifestaciones clínicas como trombosis,
abortos de repetición, vegetaciones en válvulas cardiacas y trombocitopenia. Esta la embolia de
pulmón, hipertensión pulmonar, ACV, infarto intestinal o hipertensión renovascular.
MORFOLOGIA DE LA TROMBOSIS.
Los trombos se pueden desarrollar en cualquier parte del aparato cardiovascular y varian en forma y
tamaño. Los cardiacos y arteriales suelen comenzar en áreas de turbulencia o lesión endotelial, los
venosos generalmente en lugares de estasis. Los arteriales suelen crecer en forma retrograda desde el
sitio de lesión, los venosos en dirección del flujo sanguíneo, por eso ambos se propagan hacia el corazón.
El trombo es susceptible de fragmentarse y causar embolias.
Los trombos tienen líneas de Zahn, laminaciones apreciables a simple vista, que son depósitos claros de
plaquetas y fibrina alternando con capas más oscuras con numerosos eritrocitos. Esas laminaciones
indican que se formó en sangre que fluía, pudiendo distinguir coágulos pre muerte o pos muerte.
Trombos murales: se producen en cavidades cardiacas o la luz de la aorta. Los cardiacos son
promovidos por alteraciones de la contracción del miocardio o lesiones endomiocardicas. Los aórticos son
promovidos por placas ateroescleróticas ulceradas y dilataciones aneurismáticas.
Trombos arteriales: son a menudo oclusivos, mas frecuentes en coronarias, cerebrales y femorales.
Habitualmente están superpuestos a una placa ateroesclerótica rota, también puede ser por otras lesiones
vasculares.
Trombosis venosa (flebotrombosis) es casi siempre oclusiva y el trombo forma un largo molde de la luz.
Suelen tener más eritrocitos y pocas plaquetas, por eso se llaman trombos rojos o de estasis. Son
firmes, unidos a la pared del vaso y tienen líneas de Zahn. Las venas de las extremidades inferiores son
las mas afectadas.
Coagulos post mortem: gelatinosos y tienen una porción inferior roja oscura, donde se depositaron
eritrocitos, y una porción superior de color amarillo “grasa de pollo”.
Vegetaciones: trombos sobre las válvulas cardiacas. Bacterias y hongos transportados en la sangre son
capaces de adherirse a válvulas previamente dañadas, la alteración del flujo sanguíneo y lesión endotelial
inducen la formación de grandes masas tromboticas (endocarditis infecciosa). Otras vegetaciones
esteriles sobre válvulas no infectadas en personas con hipercoagulabilidad (endocarditis tromboticas no
bacterianas). Endocarditis verrugosa esteril (endocarditis de Libman-Sacks) en lupus eritematoso
sistémico.
EVOLUCION DEL TROMBO.
Si se sobrevive a la trombosis inicial, los trombos pueden pasar por los siguientes procesos.
Propagacion: acumulan mas plaquetas y fibrina.
Embolia: se despreden y migran a otros lugares de la vasculatura.
Disolucion: por fibrinólisis, produciendo la contracción rápida y desaparición total de trombos
recientes.
Organización y recanalización: con el tiempo se forman canales capilares que reestablecen la
continuidad de la luz original.
CARACTERÍSTICAS CLINICAS.
Son motivos de atención cuando obstruyen arterias o venas o generan embolos. La presentación depende
de la zona afectada. Los venosos pueden ocasionar congestion dolorosa y edema distal a la obstrucción,
son preocupantes por la tendencia a generar embolia de pulmón. Los arteriales tienen su problema clínico
principal con la oclusión de un vaso importante.

Trombosis venosa: se producen en mayoría en venas de la pierna. Los superficiales aparecen en las
safenas, causan congestion local, tumefacción, dolor y sensibilidad pero casi nunca producen embolias. La
trombosis venosa profunda es mas grave porque se desplazan con frecuencia a los pulmones provocando
infartos pulmonares, en 50% de los casos son asintomáticas. Las TVP se asocian a estados de
hipercoagulabilidad, factores predisponentes son inmovilización, reposo e ICC, traumatismos, cirugía,
quemaduras.
Trombosis arteriales y cardiacas. Como causa fundamental la ateroesclerosis, asociada a perdida de
integridad endotelial y flujo sanguíneo anómalo. El IAM predispone a los trombos murales. Los trombos
murales cardiacos y aórticos tienden a causar embolias, con probabilidad en encéfalo, riñones y bazo.
Coagulacion intravascular diseminada.
No es una enfermedad especifica, sino la complicación de trastornos asociados con activación sistémica
de la trombina. La CID provoca la formación diseminada de trombos en la microcirculación, los cuales
pueden causar insuficiencia circulatoria difusa y disfunción de órganos, especialmente encéfalo, pulmones,
corazón y riñon.
Embolia.
Un embolo es una masa intravascular desprendida, solida, liquida o gaseosa, transportada por la sangre
desde su punto de origen a un lugar distinto donde causa disfunción o infarto tisular. La mayoría de
émbolos son trombos desprendidos. Otros pueden ser gotitas de grasa, burbujas de nitrógeno, desechos
ateroescleroticos, fragmetos de tumor, medula osea o incluso cuerpos extraños. Los embolos se
desplazan por la sangre hasta que encuentran vasos demasiado pequeños para permitir su paso y causan
una oclusión vascular parcial o completa.
EMBOLIA DE PULMÓN
Se originan en trombosis venosas profundas y son la forma mas frecuente. El 95% tienen origen en
trombosis venosa profunda de las piernas. Los trombos fragmentados de TVP son transportados hasta el
corazón derecho y luego se atascan en las arterias pulmonares, puede disponerse en la arteria pulmonar
en si, en su bifurcación (embolia en silla de montar) o en ramificaciones mas pequeñas. Principales
consecuencias:
La mayoría son silientes por su pequeño tamaño.
Si obstruyen el 60% o mas de la circulación pulmonar puede darse muerte súbita, IC derecha o
colapso cardiovascular.
La obstrucción de arterias de tamaño mediano con la rotura vascular consiguiente puede provocar
hemorragia pulmonar.
La obstrucción de arteriolas pulmonares pequeñas y terminales produce hemorragia o infarto.
Multiples embolos pueden causar hipertensión pulmonar e insuficiencia del ventrículo derecho
TROMOEMBOLIA SISTÉMICA.
La mayoría de los embolos sistémicos provienen de trombos murales intracardiacos, asociados a la pared
del VI y a la dilatación de la AI y fibrilación auricular. El resto tiene origen en aneurismas aórticos, placas
ateroescleróticas, vegetaciones valvulares y trombos venosos. A diferencia de los venosos que van al
pulmon, estos embolos arteriales se desplazan a muy distintas zonas, la mayoría termina alojándose en
extremidades inferiores o encéfalo. Las consecuencias dependen de la vulnerabilidad de los tejidos a
isquemia, el calibre del vaso ocluido, la presencia de vascularización colateral, pero por lo general causan
infarto tisular.
EMBOLIA GRASA Y MEDULAR.
Se puede encontrar globulos de grasa microscópicos en la vasculatura pulmonar tras fracturas de huesos
largos o en traumatismos de partes blandas y quemaduras. Estas lesiones rompen sinusoides vasculares
permitiendo que la medula o tejido adiposos se introduzcan en los vasos y alcancen los pulmones.
Sindrome de embolia grasa es el termino usado a la minoría de pacientes que llega a estar sintomático, se
caracteriza por: insuficiencia pulmonar, síntomas neurológicos, anemia y trombocitopenia.
EMBOLIA GASEOSA.
Las burbujas de gas en la circulación se pueden unir y formar masas espumosas que obstruyen el flujo
vascular y causan lesiones isquémicas distales. Una forma de embolia gaseosa, el síndrome de
descompresión, se produce cuando una persona experimenta un descenso brusco de la presión
atmosférica. La formación rápida de burbujas de gas en los musculos esqueléticos y tejidos de soporte de
articulaciones es responsable de la aeroembolia o enfermedad por descompresión.
EMBOLIA DE LIQUIDO AMNIÓTICO.
Es la 5ta causa de mortalidad materna en todo el mundo, provoca trastornos neurológicos permanentes en
85% de las sobrevivientes. Es una complicación del parto y posparto inmediato. Se caracteriza por disnea
brusca, cianosis y shock, seguidos de alteraciones nerologicas como cefalea, convulsiones y coma. Si
sobrevive se suele desarrolla edema de pulmón acompañado de coagulación intravascular diseminada. La
causa es la llegada de liquido amniótico o tejido fetal a la circulación materna a través de un desgarro de
las membranas placentarias o rotura de venas uterinas.
Infarto.
Un infarto es un área de necrosis isquémica causada por oclusión de la vascularización arterial o el
drenaje venoso. En la mayoría hay trombosis o embolia arterial. Otras causas menos frecuentes son
vasoespasmo local, hemorragia en una placa ateromatosa o compresión extrínseca de vasos.
Factores que influyen en el desarrollo del infarto.
Anatomia de la vascularización. La existencia de una irrigación sanguínea alternativa es el
determinante mas importante de la aparición o no de lesiones tisulares en oclusiones vasculares.
Por ejemplo pulmones, hígado, mano, antebrazo tienen doble vascularización y son menos
sensibles, no siendo asi la circulación renal o esplénica.
Velocidad de la oclusión. Las de desarrollo lento tienen menos probabilidad de causar infarto
porque dejan tiempo para la creación de vías colaterales de perfusión.
Vulnerabilidad del tejido a la hipoxia. Las neuronas sufren daños irreversibles a los 3-4 minutos, las
miocárdicas también son bastante sensibles, a los 20-30 min. Los fibroblastos del miocardio son
viables tras muchas horas de isquemia.
Hipoxemia: una concentración baja de O2 aumenta la probabilidad de infarto.
MORFOLOGIA DEL INFARTO.
Se clasifican según su color y la presencia o ausencia de infección: son rojos (hemorrágicos) o blancos
(anémicos), y sépticos o estériles.
Infarto rojo
Infarto blanco
Oclusiones venosas
Oclusiones arteriales
Tejidos laxos y esponjosos (pulmón)
Tejidos solidos
Se puede acumular sangre en la zona infartada
No se acumula sangre en el área infartada
En tejidos de doble circulación (pulmón, intestino,
hígado)
Tejidos de circulación terminal (corazón, riñon,
bazo)
En tejidos previamente congestionados con
drenaje venoso lento
Cuando se restablece el flujo en una zona con
oclusión arterial previa.
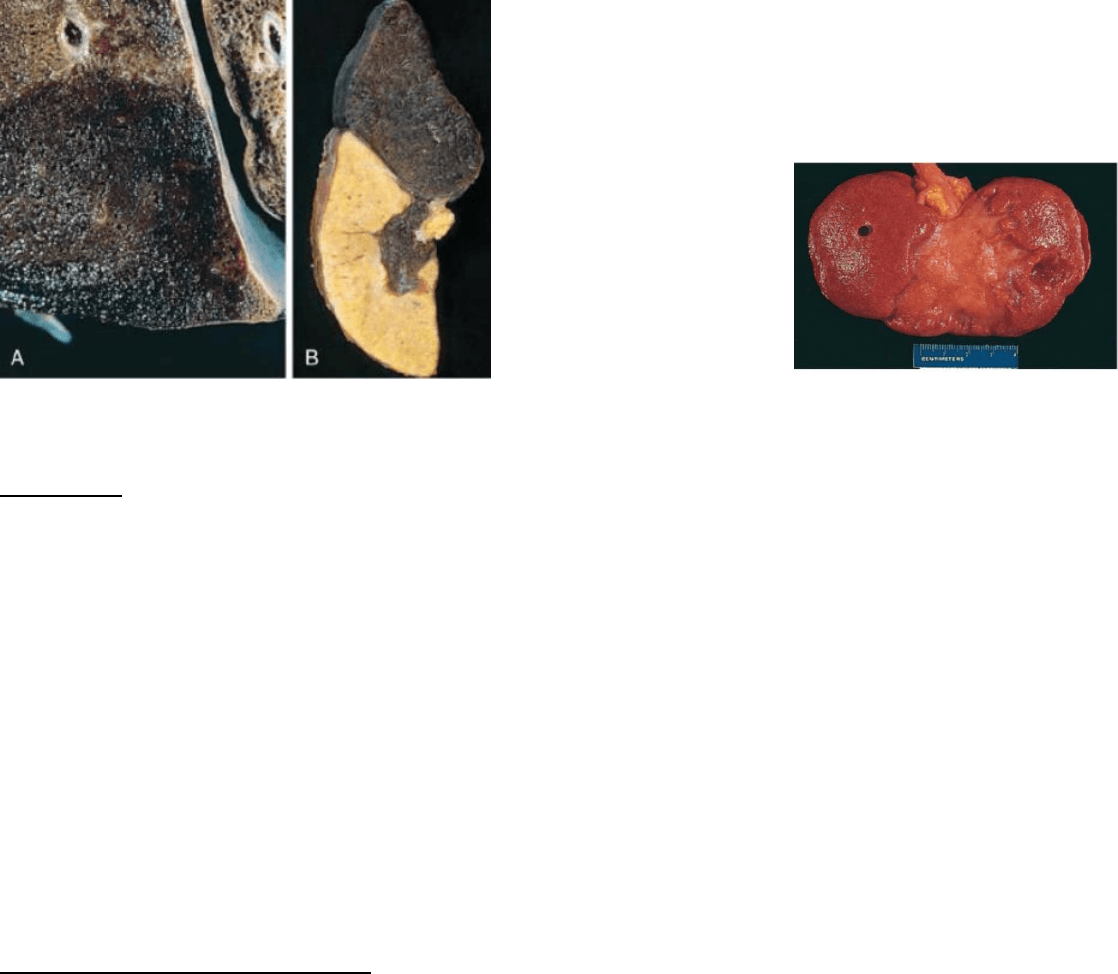
Tienden a adoptar forma de cuña, con el vaso ocluido en el vértice y base formada por la periferia del
órgano. Si la base es una superficie serosa puede haber exudado fibrinoso por reacción inflamatoria aguda
a los mediadores liberados por celulas dañadas y necróticas.
Los infartos recientes están mal definidos y son ligeramente hemorrágicos, a los días los bordes se definen
mejor.
Los infartos secundarios a oclusiones arteriales en órganos con vascularización doble son mas claros y
mejor definidos.
En el pulmón los infartos hemorrágicos son la norma. En ellos los eritrocitos extravasados son fagocitados
por macrófagos, que convierten el hierro del hemo en hemosiderina, por eso a veces una hemorragia
extensa puede dejar un residuo firme y marron rico en hemosiderina.
La característica histológica dominante del infarto es la necrosis coagulativa isquémica. La mayoría de
los infartos son reemplazados en ultimo termino por una cicatriz. El encéfalo es la excepción a la norma,
ya que los infartos del SNC resultan en necrosis licuefactiva.
Los infartos sépticos se producen por embolia de vegetaciones infectadas de válvulas cardiacas o cuando
los microbios colonizan el tejido necrótico. En estos casos el infarto se convierte en un absceso.
Infarto rojo pulmonar hemorrágico en forma de cuña /
infarto blanco bien definido en el bazo / infarto renal
reemplazado por una cicatriz fibrosa.
Shock.
El shock es un estado en el que un gasto cardiaco reducido o la disminución del volumen sanguíneo
circulante eficaz alteran la perfusión tisular y provocan hipoxia celular. En el inicio es una lesión reversible,
pero si es prolongado es irreversible y mortal. Sus causas son:
Shock cardiogenico: por disminución de GC por insuficiencia de la bomba miocárdica. Puede ser
por infarto, arritmias, taponamiento cardiaco y embolia pulmonar.
Shock hipovolémico: por disminución de GC por un volumen sanguíneo reducido, como en
hemorragias masivas o pérdidas de líquidos por quemaduras graves.
Shock asociado a inflamación sistémica: desencadenado por diferentes agresiones como
infecciones microbianas, quemaduras, traumatismos, pancreatitis. Es común una oleada de
mediadores inflamatorios que producen vasodilatación arterial, extravasación y remanso de sangre
venosa.
Con menos frecuencia aparecen shock neurogeno (accidentes anestésicos o lesiones de la medula
espinal), anafiláctico (reacciones de hipersensibilidad).
PATOGENIA DEL SHOCK SÉPTICO.

Es el shock causado por infecciones microbianas. Es la primer causa de muerte en cuidados intensivos.
Es producido en mayor frecuencia por infecciones bacterianas por gram +, en segundo lugar gram -, y
hongos en tercer lugar. Distintos elementos microbianos pueden desencadenar el proceso. Celulas del
sistema inmune reconocen varias sustancias derivadas de los microorganismos y luego son activadas.
Una vez activadas ponen en marcha rtas inflamatorias. Los factores principales de la fisiopatología del
shock séptico son:
Respuestas inflamatorias y antiinflamatorias. Elementos de la pared celular microbiana se unen a
receptores del sistema inmune desencadenando rtas proinflamatoias. Las vías asociadas a TLR
son las involucradas. Al ser activadas las celulas inmunes producen diferentes citosinas y
mediadores, también se generan ERO y lípidos mediadores como PG y PAF. Estos mediadores
inducen al endotelio a regular a la alza la expresión de moléculas de adhesión, estimulando aun
mas la producción de citosinas y quimiocinas. Se activa el complemento, contribuyendo al estado
inflamatorio. Tambien se activa la via de coagulación. Este estado hiperinflamatorio
desencadenado por la sepsis activa inmunodepresores contrarreguladores. Como resultado, los
pacientes en sepsis oscilan entre estados de hiperinflamacion e inmunosupresión.
Activacion y lesión del endotelio. Provoca extravasación vascular generalizada y edema tisular. Un
efecto de las citosinas es distender las uniones herméticas entre celulas endoteliales, haciendo que
salga el contenido de los vasos, con una acumulación de edema rico en proteinas por todo el
orgnismo. Esto impide la perfusión tisular. El endotelio activado produce NO y otros mediadores
inflamatorios que contribuyen a la vasodilatación e hipotensión sistémica.
Inducción de un estado procoagulante. La extravasación vascular y edema tisular reducen el flujo
sanguíneo produciendo estasis, estos efectos provocan la activación de trombina y el deposito de
trombos en los pequeños vasos por todo el organismo (coagulación intravascular diesminada)
comprometiendo mas la perfusión tisular.
Anomalias metabólicas. Los pacientes sépticos presentan resistencia a insulina e hiperglucemia, la
cual reduce la acción de los neutrofilos suprimiendo su capacidad bactericida.
Disfuncion de órganos. La hipotensión sistémica, el edema intersticial y la trombosis de pequeños
vasos reducen el aporte de oxigeno y nutrientes a los tejidos. Las concentraciones elevadas de
citosinas y mediadores reducen la contractilidad miocárdica y el GC, y la mayor permeabilidad
vascular y lesión endotelial pueden provocar el síndrome de dificultad respiratoria aguda. Estos
factores se unen para causar la insuficiencia de multiples órganos, como riñones, hígado,
pulmones y corazón, que termina en muerte.
FASES DEL SHOCK.
Se conocen las del shock hipovolémico y cardiogenico. A menos que la lesión sea masiva y rápidamente
mortal como la rotura de un aneurisma aórtico, estos tipos de shock evolucionan en tres fases generales:
Fase no progresiva inicial: activación de mecanismos compensadores reflejos y mantención de la
perfusión de órganos vitales.
Fase progresiva: hipoperfusion tisular, deterioro circulatorio y desequilibrios metabólicos.
Fase irreversible: cuando se sufrieron lesiones celulares y tisulares tan graves que la supervivencia
es imposible.
Al inicio mecanismos neurohumorales ayudan a mantener el GC y PA, como reflejos barorreceptores,
liberación de catecolaminas y ADH, activación del SRAA y estimulación simpática generalizada. El efecto
es: taquicardia, vasoconstricción y conservación renal de liquido. En la fase progresiva se produce hipoxia
tisular generalizada. Ante falta de oxigeno se pasa a una respiración celular anarobica con producción
excesiva de acido láctico, que reduce el pH tisular y amortigua la rta vasomotora; las arteriolas se dilata y
la sangre se remansa en la circulación, empeorando el GC. Ante la hipoxia generalizada los órganos
vitales se ven afectados y comienzan a fallar. En la fase irreversible hay salida de enzimas lisosómicas.
CONSECUENCIAS CLINICAS.

Dependen de la lesión causal.
Hipovolemico y cardiogenico: hipotensión, pulso rápido y débil, taquipnea, piel cianótica fría y humeda.
Septico: incialmente la piel caliente y enrojecida por vasodilatación periférica. Luego provoca disfunción
cardiaca, cerebral y pulmonar, alteraciones electrolíticas y acidosis metabolica. Puede verse insuficiencia
renal con disminución de la diuresis. Se ven compliaciones por coagulopatias.
MORFOLOGÍA.
Cambios tisulares y celulares en shock cardiogenico e hipovolémico: correspondientes a la lesión hipoxica,
cambios patentes en el encéfalo, corazón, pulmones, riñones, suprarrenales y tubo digestivo. Cambios
suprarrenales: depleción de lípidos en las celulas corticales. Los riñones presentan necrosis tubular aguda.
Los pulmones casi nunca se afectan en el shock hipovolémico, pero si es causado por sepsis o
traumatismos puede desarrollar lesión alveolar difusa. En el shock séptico la CID provoca deposito
generalizado de microtrombos en encéfalo, corazón, pulmones, riñon, suprarrenales y tubo digestivo. El
consumo de plaquetas produce aparición de petequias en la piel.

trans. metabolicos.pdf
 Estamos procesando este archivo...
Estamos procesando este archivo...
 Lamentablemente la previsualización de este archivo no está disponible. De todas maneras puedes descargarlo y ver si te es útil.
Lamentablemente la previsualización de este archivo no está disponible. De todas maneras puedes descargarlo y ver si te es útil.
Descargar
Estamos procesando este archivo...
Lamentablemente la previsualización de este archivo no está disponible. De todas maneras puedes descargarlo y ver si te es útil.