
Sanchez, Nahuel
Apunte de Genética
2do Año
Sanchez, Nahuel 1er ERA
+5555991582459 @gabriel_apuntes
MUTACIONES
Es el cambio en la secuencia de un nucleótido o en la organización del ADN de un ser
vivo, lo que produce una variación en las características de este y no necesariamente se
transmite a la descendencia.
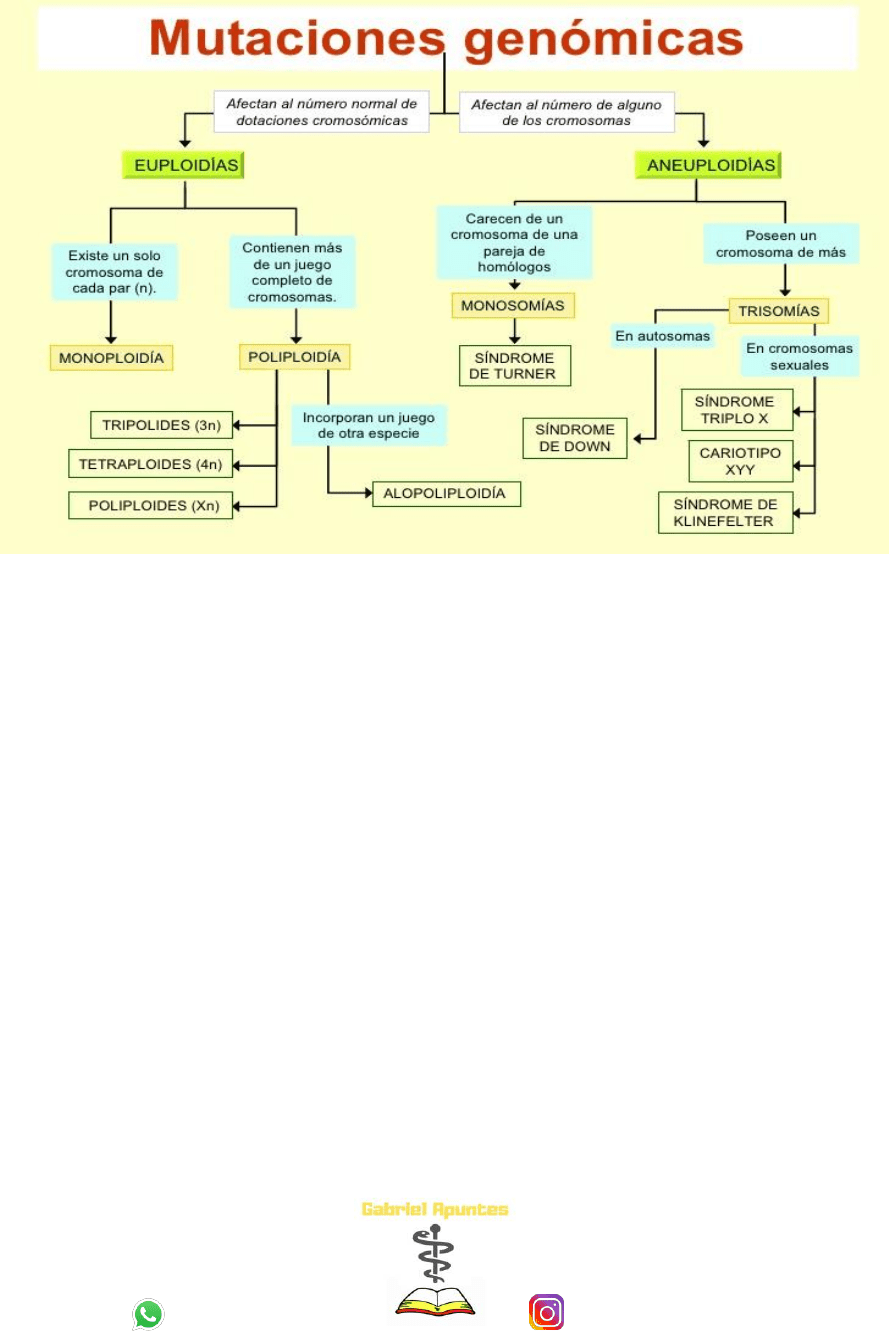
EUPLOIDIAS: es la variación en el número de cromosomas
a) Monoploidia: presenta solo un juego de cromosomas. (23 cromosomas).
b) Poliploidia: presenta más de dos juegos de cromosomas.
Triploides = 69 cromosomas
Tetraploides = 92 cromosomas
ANEUPLOIDIAS: se dan cuando está afectada solo una parte del juego cromosómico y el
cigoto presenta cromosomas de más o de menos.
a) Monosomia (1 cromosoma menos), EJ.: SINDROME DE TURNER
b) Trisomías (1 cromosoma mas), EJS:

Sanchez, Nahuel 1er ERA
+5555991582459 @gabriel_apuntes
1- SINDROME DE DOWN (trisomía del cromosoma 21): es la causa más frecuente de
retraso mental identificable de origen genético. Tiene una incidencia de 1 cada 700 recién
nacidos vivos, y la posibilidad aumenta con la edad materna.
Causa: presencia de una copia extra del cromosoma 21.
Características clínicas
Retardo mental en todos los casos
Alta probabilidad de tener cardiopatías congénitas
Micrognatia
Ojos almendrados con manchas de Brushfield en el iris
Manos pequeñas y cuadradas
Criptorquidia frecuente
Hipotiroidismo
Problemas visuales
Predisposición al sobre peso por la baja estatura
Diagnostico
Cariotipo
Características fenotípicas
Expectativa de vida: alrededor de 60 años
2- SINDROME DE PATAU (trisomía del cromosoma 13): es la trisomía menos frecuente,
presenta mayor frecuencia en mujeres que en varones y origina graves cuadros de retraso
mental y una bajísima expectativa de vida. Tiene una incidencia de 1 cada 10.000 recien
nacidos vivos, y la posibilidad aumenta con la edad materna.
Causa: presencia de una copia extra del cromosoma 13.
Características clínicas
Retraso mental
Frente aplanada
Labio leporino con fisura palatina
Polidactilia
Cardiopatías congénitas
Diagnostico
Cariotipo
Expectativa de vida: casi todos mueren antes del año de vida

Sanchez, Nahuel 1er ERA
+5555991582459 @gabriel_apuntes
3- SINDROME DE EDWARDS (trisomía del cromosoma 18): es la segunda trisomía mas
frecuente, se presenta con mayor frecuencia en mujeres que en varones y origina graves
cuadros de retraso mental y una bajísima expectativa de vida. Tiene una incidencia de 1
cada 6.000 recien nacidos vivos, y la posibilidad aumenta con la edad materna.
Causa: presencia de una copia extra del cromosoma 18.
Características clínicas
Cardiopatías congénitas
Retraso mental
Micrognatia
Manos trisomicas
Microcefalia
Pies en mecedora
Diagnostico
Cariotipo
Expectativa de vida: cerca del 50% mueren en las primeras semanas de vida, y
solo el 5% sobrevive hasta el año.
SINDROME DE CRI DU CHAT O DEL MAULLIDO DE GATO
Se produce por la pérdida (deleción) del brazo corto del cromosoma 5, lo que se
denomina deleción y que es una de las posibles aberraciones cromosómicas estructurales,
tiene una incidencia de 1 cada 20.000 o 50.000 recién nacidos vivos y es más frecuente
en mujeres que en varones.
El síntoma más característico de este síndrome es un llanto semejante al maullido de
un gato, originado por las malformaciones del aparato respiratorio.
Características clínicas
Retraso mental
Bajo peso al nacer
Llanto característico (maullido de gato)
Microcefalia
Micrognatia
Sindáctila en manos y pies
Cardiopatías congénitas
Malformaciones del aparato respiratorio
Diagnostico
Cariotipo
Características fenotípicas
Expectativa de vida: esta disminuida, aunque la mayoría alcanza la edad adulta
(alrededor de los 50 años)

Sanchez, Nahuel 1er ERA
+5555991582459 @gabriel_apuntes
TIPOS DE HERENCIA
Existen distintos patrones de herencia, esto es, distintas formas de transmitir rasgos o
enfermedades, cuya información se encuentra presente en nuestros genes, dependiendo
de la ubicación precisa del gen en cuestión.
Esto determina que según la ubicación del gen que posea la información de un rasgo o
enfermedad, deberemos considerar cuatro patrones de herencia:
Herencia autosómica
Herencia ligada al cromosoma X
Herencia ligada al cromosoma Y
Herencia mitocondrial
HERENCIA AUTOSOMICA: se denomina de esta manera a todo tipo de información que
se transmita a través de los cromosomas somáticos o autosomas, es decir, del par 1 al 22.
Podemos clasificarla en:
1- HERENCIA AUTOSOMICA DOMINANTE: decimos que este tipo de herencia es
dominante cuando es suficiente heredar de uno de los progenitores el rasgo en
cuestión para que exprese en el fenotipo, por lo tanto, se expresará tanto en
individuos homocigotos como heterocigotos.
ACONDROPLASIA
Es un trastorno genético que se transmite por un patrón de herencia autosómico
dominante y consiste en un anormal desarrollo del proceso de osificación endocondral
(propio de los huesos largos), lo cual ocasiona una de las distintas formas posibles de
enanismo.
Esta enfermedad tiene una incidencia de 1 cada 25.000 recién nacidos vivos
Causa: mutación en el gen FGFR3, el cual se ubica en el brazo corto del
cromosoma 4.
Características clínicas
Escaso desarrollo de los huesos largos
Altura del tronco es normal
Manos cortas y anchas, con dedos rechonchos
Patologías asociadas
Retraso motor al nacer
Hiperlaxitud de las rodillas
Lordosis
Cifosis
Estenosis del canal medular y foramen magno
Diagnóstico: las características clínicas evidentes alcanzan para hacer el
diagnóstico de la enfermedad

Sanchez, Nahuel 1er ERA
+5555991582459 @gabriel_apuntes
Tratamiento: la acondroplasia no posee un tratamiento curativo, sino paliativo de
las complicaciones que pueden derivar de las patologías asociadas a la enfermedad.
Expectativa de vida: es igual a la de cualquier persona sana.
2- HERENCIA AUTOSOMICA RECESIVA: decimos que este tipo de herencia es
recesiva cuando es necesario heredar de ambos progenitores el rasgo en cuestión,
por lo tanto, para que ese exprese en el fenotipo, el individuo debe ser homocigoto
para dicho rasgo.
FIBROSIS QUISTICA (mucoviscidosis)
Muco = moco
Viscidosis = pegajoso
Es un trastorno genético que se transmite por un patrón de herencia autosómico recesivo
y que da lugar a una enfermedad crónica y progresiva que afecta a diversos sistemas y
órganos del cuerpo.
Es la enfermedad genética mortal más frecuente en la raza blanca, con una
incidencia de 1 cada 3.000 recién nacidos vivos.
Causa: mutación en el gen CFTR, el cual se ubica en el brazo largo del cromosoma
7.
Fisiopatología: la mutación descripta origina que el canal de cloro no funcione
adecuadamente, y esto tiene como consecuencia principal un espesamiento de las
secreciones exocrinas por una disminución de la cantidad de agua en las mismas.
Signos y síntomas respiratorios: la mucosidad espesa y pegajosa obstruye las
vías respiratorias, provocando:
Tos persistente con mucosidad espesa
Silbido al respirar
Falta de aliento
Intolerancia al ejercicio
Infecciones pulmonares
Congestión nasal
Signos y síntomas digestivos: la mucosidad espesa también obstruye las vías que
transportan las enzimas digestivas desde el páncreas hasta el intestino delgado.
Sin estas encimas, el intestino no puede absorber por completo los nutrientes
ingeridos, lo que ocasiona:
Heces malolientes y grasosas
Problemas para aumentar de peso y crecer
Obstrucción intestinal
Estreñimiento grave

Sanchez, Nahuel 1er ERA
+5555991582459 @gabriel_apuntes
Diagnóstico: está basado en la determinación de por lo menos 2-3 determinantes
positivos de electrolitos en sudor (se seca en forma de hoja de helecho), junto con
los siguientes, criterios clínicos:
Historia familiar de fibrosis quística
Insuficiencia pancreática exocrina
Enfermedad pulmonar obstructiva crónica (EPOC)
Síndrome de perdida de sal (piel salada)
Tratamiento: la fibrosis quística no tiene un tratamiento curativo, sino preventivo
y paliativo.
Cuando la enfermedad llega a etapas muy avanzadas con empeoramiento severo de
la función respiratoria, se debe recurrir a un transplante de pulmón como única
medida efectiva para salvar la vida del paciente.
Expectativa de vida: alrededor de 30 años.

Sanchez, Nahuel 2da ERA
+5555991582459 @gabriel_apuntes
SINDROME DE TURNER
Se trata de una mutación genómica aneuploidia, y dentro de estas, es una Monosomía,
es decir hay un cromosoma X menos. Afecta solo a mujeres.
CARIOTIPO: 45 X
Incidencia: 1 cada 2000 mujeres recién nacidas vivas; y no tiene relación con la
edad materna.
Características clínicas
Baja estatura
Cuello corto y alado
Edemas de manos y pies
Amenorrea
Esterilidad
Obesidad
Mayor riesgo de luxación congénita de cadera
Osteosporosis
Diabetes tipo II
Escaso desarrollo mamario
Estenosis de válvula aortica
Atrofia de los ovarios, por lo que hay ausencia de caracteres sexuales secundarios
Diagnostico
Observación de características clínicas
Cariotipo (45X)
Tratamiento: se realiza un tratamiento con estrógenos para que se desarrollen los
caracteres sexuales secundarios.
SINDROME DE LA SUPERHEMBRA O TRIPLE X
Se trata de una mutacion genómica aneuploidia, y dentro de estas es una trisomía en
cromosomas sexuales, es decir hay un cromosoma X de mas. Afecta solo a mujeres.
NO HAY ESTERILIDAD NI INFERTILIDAD
CARIOTIPO: 47 XXX
Incidencia: 1 cada 1500 mujeres recién nacidas vivas, y no tiene relacion con la
edad materna.
Características clinicas
Talla alta (altura elevada)
Menopausia precoz
Problemas de aprendizaje
Dislexia
Problemas del habla y lenguaje

Sanchez, Nahuel 2da ERA
+5555991582459 @gabriel_apuntes
La mayoría de las mujeres con este síndrome tiene desarrollo sexual normal y son
capaces de concebir niños.
SINDROME DE KLINEFELTER
Se trata de una mutación genómica aneuploidia, y dentro de estas es una trisomía en
cromosomas sexuales, es decir hay un cromosoma X de más. Afecta solo a hombres.
CARIOTIPO: 47 XXY
Incidencia: 1 cada 1000 varones recién nacidos vivos, y tiene relación con la edad
materna (mayor riesgo en madres mayores de 35 años).
Características clínicas
Atrofia testicular, por lo que hay ausencia de caracteres sexuales secundarios
Altos y delgados
Ginecomastia
Esterilidad
Poca masa muscular
Micropene
Criptorquidia
Cuerpo afeminado, debido a la falta de testosterona
Diagnostico
Observación de características clínicas
Cariotipo (47 XXY)
Tratamiento: se realiza un tratamiento con testosterona para que se desarrollen
los caracteres sexuales secundarios.
SINDROME DEL SUPERMACHO O DE JACOBS
Se trata de una mutación genómica aneuploidia, y dentro de estas es una trisomía en
cromosomas sexuales, es decir hay un cromosoma Y de más. Afecta solo a varones.
NO HAY ESTERILIDAD
CARIOTIPO: 47 XYY (cariotipo de la criminalidad)
características clínicas
altos y delgados
acné
manos y pies grandes
agresivos
antisociales
no diferencian el bien del mal

Sanchez, Nahuel 2da ERA
+5555991582459 @gabriel_apuntes
SINDROME DE MARFAN
Se trata de una enfermedad genetica de herencia autosómica dominante, del tejido
conectivo y que afecta principalmente al sistema cardiovascular, ocular y musculo
esquelético.
Se caracteriza por una alta penetrancia (cercana al 100%).
Incidencia: 1 cada 5.000 personas
Causa: mutaciones del gen FBN1 en cromosoma 15. Este gen codifica la fibrilina
que es un componente esencial de las fibras elásticas.
Fisiopatología: hay alteración de las fibras elásticas, por lo que se ven afectados
los órganos que son ricos en este tipo de fibras.
Características clínicas
Alteraciones esqueléticas
Paciente alto, con extremidades largas y aranodactilia
Hiperlaxitud articular
Malformaciones de columna vertebral: * cifosis
* escoliosis
Deformidades del tórax: * pectum excavatum (pecho hundido)
* pectum carinatum (pecho de paloma)
Alteraciones oculares
Ectopia lentis
Alteraciones cardiovasculares: causan la muerte en el 45% de los casos
Prolapso de la válvula mitral
Dilatación de la aorta ascendente, con necrosis quística de la capa media
OSTEOGENESIS IMPERFECTA
Se trata de una enfermedad genética de herencia autosómica dominante, causada por
alteraciones en la formación del colágeno tipo I.
Presenta fragilidad ósea con un espectro variable de manifestaciones clínicas que
pueden ir desde algunas fracturas hasta deformidades Oseas severas.
Incidencia: 1 cada 10.000 nacidos vivos
Causa: mutaciones en el gen COL1A1 localizado en el brazo largo del cromosoma
17; y en el gen COL1A2 localizado en el brazo largo del cromosoma 7.
Fisiopatología: debido al alto predominio de colágeno en el hueso, se produce una
desmineralización ósea anormal, pero también a otros niveles: escleróticas, piel, dientes,
oídos, etc.

Sanchez, Nahuel 2da ERA
+5555991582459 @gabriel_apuntes
Manifestaciones clínicas: compuesta por una triada patognomónica:
Fragilidad ósea
Escleróticas azules
Sordera prematura
Características clínicas principales
Deformidades esqueléticas
Deformidades en la columna vertebral
Crecimiento inadecuado de los huesos temporales y parietales
Hipoacusia conductiva
Escleróticas azules
Dentinogenesis imperfecta (inadecuada formación de esmalte con fracturas
dentarias).
Diagnostico
Examen físico enfocado al sistema óseo, incluyendo evaluación de columna
vertebral, y se debe buscar otras características típicas como ser: escleróticas azules,
odontogenesis imperfecta, etc.
Pruebas bioquímicas encaminadas a evaluar las características y la cantidad del
colágeno tipo I.
Tratamiento
Es encaminado a disminuir la reabsorción ósea y aumentar la formación de hueso,
por lo que se acude a bifosfonatos, los cuales disminuyen la acción osteoclastica y
aumentan la acción osteoblastica.
ALOPECIA ANDROGENETICA O CALVICIE
Se hereda con un patrón clínico masculino (MAGA) o femenino (FAGA), y se produce
por la acción de los andrógenos en personas con predisposición genética.
Los andrógenos, sobre todo la dihidrotestosterona, actúan sobre el folículo piloso del
cuero cabelludo atrofiándolo.
Puede empezar en cualquier momento después de la pubertad, aunque su incidencia
aumenta con la edad.

Sanchez, Nahuel 2da ERA
+5555991582459 @gabriel_apuntes
HERENCIA LIGADA AL X RECESIVA
En general solo están afectados los varones
No hay transmisión de padre a hijo
Transmisión diagonal a través de mujeres portadoras
Los hijos de un varón afectado serán todos sanos y sus hijas todas portadoras.
Los descendientes de mujeres portadoras: si son varones tendrán 50% de
probabilidad de ser afectados; si son mujeres tendrán 50% de probabilidades de ser
portadoras.
EJEMPLOS DE HERENCIA LIGADA AL X RECESIVA:
HEMOFILIA
Es la enfermedad hemorrágica hereditaria que se caracteriza por la incapacidad de
formar coágulos en presencia de heridas incluso superficiales.
CAUSAS Y TIPOS
La enfermedad está causada por la ausencia congénita de determinados factores de la
coagulación de la sangre.
Hemofilia A: es la más común y esta originada por un déficit del factor VIII.
Hemofilia B: esta originada por un déficit del factor IX.
INCIDENCIA
Hemofilia A: 1 cada 10.000 recién nacidos vivos
Hemofilia B: 1 cada 40.000 recién nacidos vivos
SIGNOS Y SINTOMAS
Hemorragias externas: epistaxis, hematuria, gastrointestinales, etc.
Hemorragias internas: intracraneanas, retroperitoneales, hemartrosis, oculares,
musculares, etc.
Hematomas espontáneos o por traumas

Sanchez, Nahuel 2da ERA
+5555991582459 @gabriel_apuntes
DIAGNOSTICO
Es un diagnostico médico, basado en el antecedente clínico de sangramiento y
exámenes de laboratorio, EJ: * tiempo parcial de tromboplastina = normal
* tiempo de protombina = normal
* cuantificación de factores (VIII y IX)
* estudios de agragacion y secreción plaquetaria
TRATAMIENTO
Concentrados heterologos: se obtienen a partir de sangre animal. Permite
administrar al paciente una cantidad de factor VIII equivalente a la contenida en 8 lts de
sangre humana, en una sola inyección de pequeño volumen.
DALTONISMOS
Es la alteración de los genes para pigmentos retinianos: PCR (pigmentos de conos
sensibles al rojo) y PCV (pigmentos de conos sensibles al verde). Ambos están ubicados en
el brazo largo del cromosoma X (Xq28).
INCIDENCIA: afecta a una 8% de los varones de raza blanca y los individuos que la
padecen se clasifican en:
PROTANOPES: cuando la sensibilidad al rojo es defectuosa
DEUTERANOPES: cuando la sensibilidad al verde es defectuosa
TRITANOPES: cuando la sensibilidad al azul es defectuosa. En el cromosoma 7 se
ubican los genes que codifican la cianopsina (pigmento para el color azul).
DIAGNOSTICO
Dentro de las pruebas realizadas sobresale el TEST DE ISHIHARA, donde en distintas
laminas con muchos puntos se representa en el fondo algunos números y figuras de
distintas tonalidades de colores primarios.
Las personas daltónicas no pueden reconocer algunos números y figuras que aparecen
en el test, o bien visualizan diferentes notaciones de acuerdo al tipo de defecto que
padezcan.

Sanchez, Nahuel 2da ERA
+5555991582459 @gabriel_apuntes
DISTROFIA MUSCULAR DE DUCHENNE
Es una enfermedad genética que afecta a 1 cada 3.600 varones recién nacidos vivos.
La mutación genética se encuentra en el cromosoma X, del cual los varones solo tiene
una copia. Los pacientes con DMD no pueden producir la proteína conocida como
distrofina, la cual es esencial para el mantenimiento de la integridad de las fibras
musculares.
Pasado el tiempo, los pacientes con este defecto sufren deterioro gradual de los
músculos, lo cual lleva a la parálisis y finalmente la muerte, normalmente cerca de los 25
años.
SINTOMAS MAS COMUNES
Retraso psicomotor
Debilidad muscular
Dificultad para el ejercicio físico
Caídas frecuentes
Necesita ayuda para levantarse del suelo
Pantorrillas con desarrollo exagerado
Camina con marcha de pato
Debilidad del musculo cardiaco
Disfunción respiratoria
TRATAMIENTO
No existe tratamiento curativo, sino paliativo
El tratamiento se dirige a evitar las complicaciones resultantes de la debilidad,
movilidad disminuida y las dificultades cardiacas y respiratorias.
El tratamiento puede implicar una combinación de fisioterapia, terapia
medicamentosa y cirugías.

Sanchez, Nahuel 3er ERA
+5555991582459 @gabriel_apuntes
ERRORES CONGENITOS DEL METABOLISMO
Los ECM son un grupo de enfermedades causadas por el bloqueo de un paso
metabólico. La causa del bloqueo es la mutación de genes responsables del
funcionamiento de dicho paso metabólico.
El mecanismo de herencia es, en casi el 100% de los casos, autosómico recesivo.
FISIOPATOLOGIA DE LOS ERRORES CONGENITOS DEL METABOLISMO
En ellos, una mutación determina la falla de una enzima. La falla enzimática puede
ocurrir de varias maneras:
1- La proteína no se produce, se produce cortada o en poca cantidad
2- La actividad de la enzima esta modificada o suprimida
De manera esquemática, los errores metabólicos funcionan de la siguiente manera:
A ---- enzima 1 ----- B ----- enzima 2 ----- C ------ enzima 3 ------- D ----- enzima 4 ------
E
Cualquier alteración en alguna de las enzimas que catalizan esas reacciones supondrá:
El bloqueo de la ruta metabólica
La no producción de
E
La apertura de vías metabólicas alternativas
La acumulación en la célula de un metabolito intermediario
EJEMPLOS DE ENFERMEDADES CUASADAS POR ERRORES CONGENITOS DEL
METABOLISMO:
GALACTOSEMIA
Es una enfermedad en la cual el cuerpo no puede metabolizar la galactosa. Las personas
con galactosemia son incapaces de descomponer la galactosa en lactosa (azúcar que se
encuentra en la leche) y glucosa.
Si a un bebe con galactosemia se le da leche, los derivados de la galactosa se acumularán
en el sistema y dañarán al hígado, cerebro, riñones y ojos.
Este documento contiene más páginas...
Descargar Completo
APUNTE GENETICA 1,2 Y 3 ERA.pdf
 Estamos procesando este archivo...
Estamos procesando este archivo...
 Lamentablemente la previsualización de este archivo no está disponible. De todas maneras puedes descargarlo y ver si te es útil.
Lamentablemente la previsualización de este archivo no está disponible. De todas maneras puedes descargarlo y ver si te es útil.
Descargar
Estamos procesando este archivo...
Lamentablemente la previsualización de este archivo no está disponible. De todas maneras puedes descargarlo y ver si te es útil.