Página 1 de 12
PATOLOGIA OSTEOMUSCULAR Y PARTES BLANDAS
ARTICULACIONES
Artrosis
También denominada artropatía degenerativa que se caracteriza por la erosión progresiva del cartílago articular. El termino
artrosis u osteoartritis implica un trastorno inflamatorio aunque a pesar de la presencia de células inflamatorias, se considera
que es una enfermedad intrínseca del cartílago, en la que las alteraciones bioquímicas y metabólicas en personas con
predisposición genética conducen a su degradación.
-Habitualmente oligoarticular aunque puede ser generalizada.
Puede ser:
Primaria: aparece de modo insidioso, sin una causa aparente, como fenómeno del envejecimiento.
Secundaria: Puede aparecer en personas más jóvenes con algún trastorno predisponente, como lesiones articulares
previas, deformidad congénita, o alguna enfermedad sistémica, como diabetes, ocronosis, hemocromatosis u obesidad
avanzada, aquí se denomina artrosis secundaria y afecta a menudo una o varias articulaciones predispuestas.
En la mujer afecta con más frecuencia a las rodillas y manos, y en el hombre a la cadera.
Patogenia: es una enfermedad multifactorial con componentes genéticos y ambientales. Los últimos están relacionados con el
envejecimiento y la sobrecarga, influenciados por obesidad, fuerza muscular y estabilidad, estructura y alineación articular. La
edad, porque la prevalencia aumenta de forma exponencial después de los 50 años. Los mecanismos que conducen a la misma
pueden dividirse en varias fases: 1) lesión del condrocito relacionada con el envejecimiento y con factores predisponentes, 2)
artrosis prematura en la que los condrocitos proliferan y secretan mediadores inflamatorios, colágenos, proteoglucanos y
proteasas que remodelan la matriz cartilaginosa e inician cambios inflamatorios secundarios en la sinovial y el hueso subcondral,
y 3) artrosis tardía, en la que la lesión repetitiva y la inflamación crónica conducen a perdida de condrocitos, marcada perdida de
cartílago y cambios extensos en el hueso subcondral.
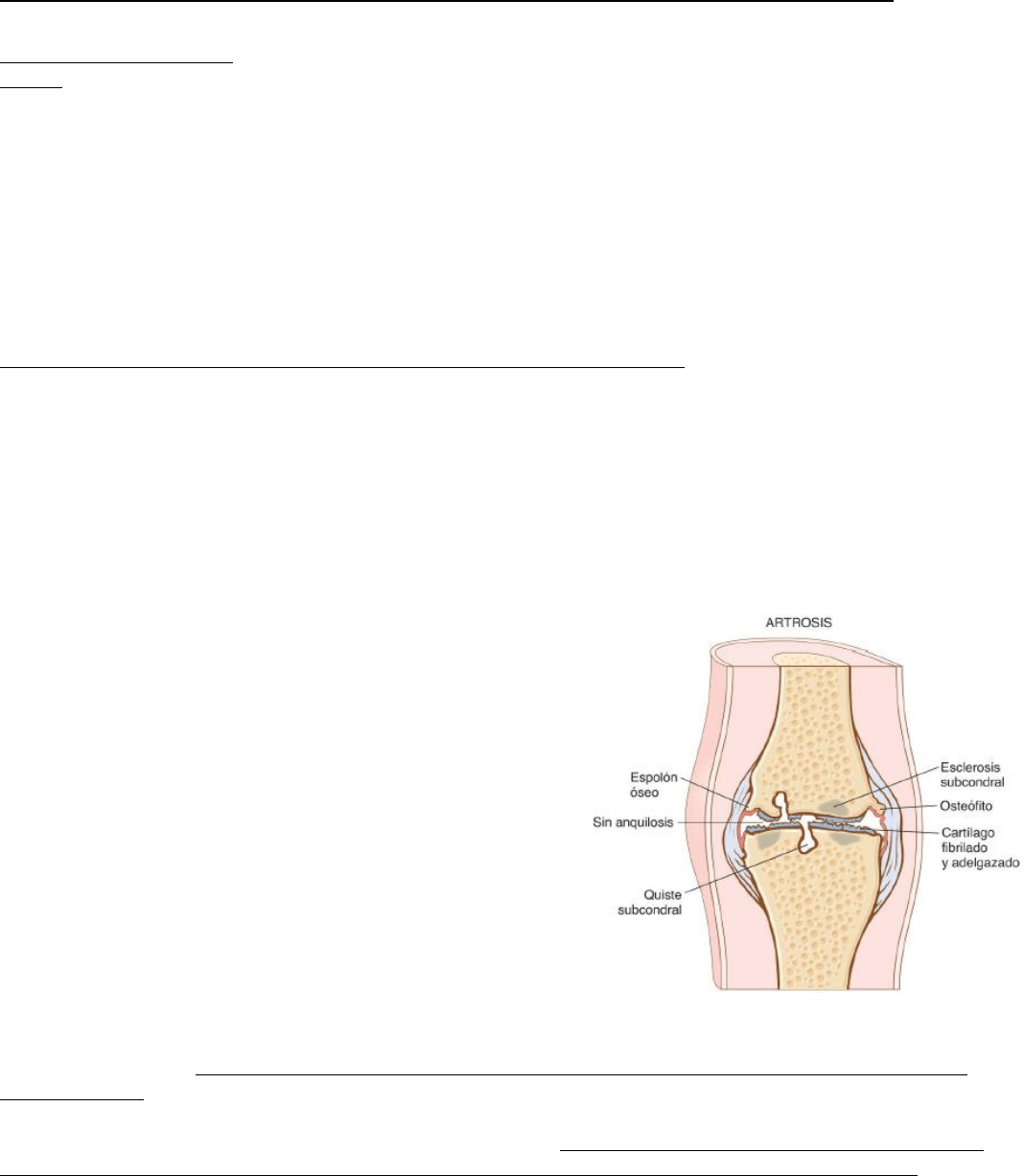
Morfología:
-En las 1ras fases, los condrocitos proliferan formando agregados,
aumenta el contenido de agua de la matriz y disminuye la concentración
de proteoglucanos. Se produce la fibrilación y figuración vertical y
horizontal de la matriz conforme se degradan las capas superficiales del
colágeno tipo 2.
-Por último, los condrocitos mueren y se desprenden porciones de
espesor total del cartílago; las piezas desprendidas entran en la
articulación formando cuerpos libres (ratones articulares).
-La lámina expuesta es la nueva superficie articular y la fricción con la
superficie opuesta alisa y endurece el hueso expuesto dándole un
aspecto de marfil pulido (eburnación ósea).
-Son frecuentes las pequeñas fracturas que permiten que el líquido
sinovial sea forzado a las regiones subcondrales cuya acumulación
aumenta de tamaño y forma quistes con una pared fibrosa.
-Aparecen osteofitos fungiformes (excresencias óseas) en los márgenes
de la superficie articular cubiertos por fibrocartílago y cartílago hialino
que se osifican gradualmente. La sinovial presenta congestión y fibrosis.
Clínica: de evolución lenta; los pacientes con artrosis primaria suelen estar asintomáticos hasta los 50 años.
-Los síntomas habituales son dolor continuo profundo que empeora con el uso, rigidez matutina, crepitación y limitación del
arco de movilidad. Los osteofitos en los agujeros intervertebrales pueden comprimir las raíces raquídeas cervicales y lumbares,
produciendo dolor, espasmos musculares, atrofia muscular y defectos neurológicos.
-Por lo general, afecta solo a una o varias articulaciones, principalmente: cadera, rodilla, vértebras lumbares y cervicales bajas,
interfalángicas proximales y distales de los dedos, carpo metacarpiana del pulgar y tarso metatarsiano del dedo gordo.
-Los nódulos de Heberden, son osteofitos prominentes en las articulaciones interfalángicas distales, son frecuentes en la mujer.
-Suele respetar la muñeca, codo y hombro.
Página 2 de 12
Artritis Reumatoide
-“Es una enfermedad inflamatoria crónica de origen autoinmunitario, que puede afectar a muchos tejidos y órganos (piel, vasos
sanguíneos, corazón, pulmones y músculos), aunque afecta principalmente a las articulaciones con una sinovitis inflamatoria y
proliferativa no supurativa que a menudo progresa a destrucción del cartílago articular y genera anquilosis articular
-Es más frecuente en las mujeres y aparece a la edad de 40-70 años
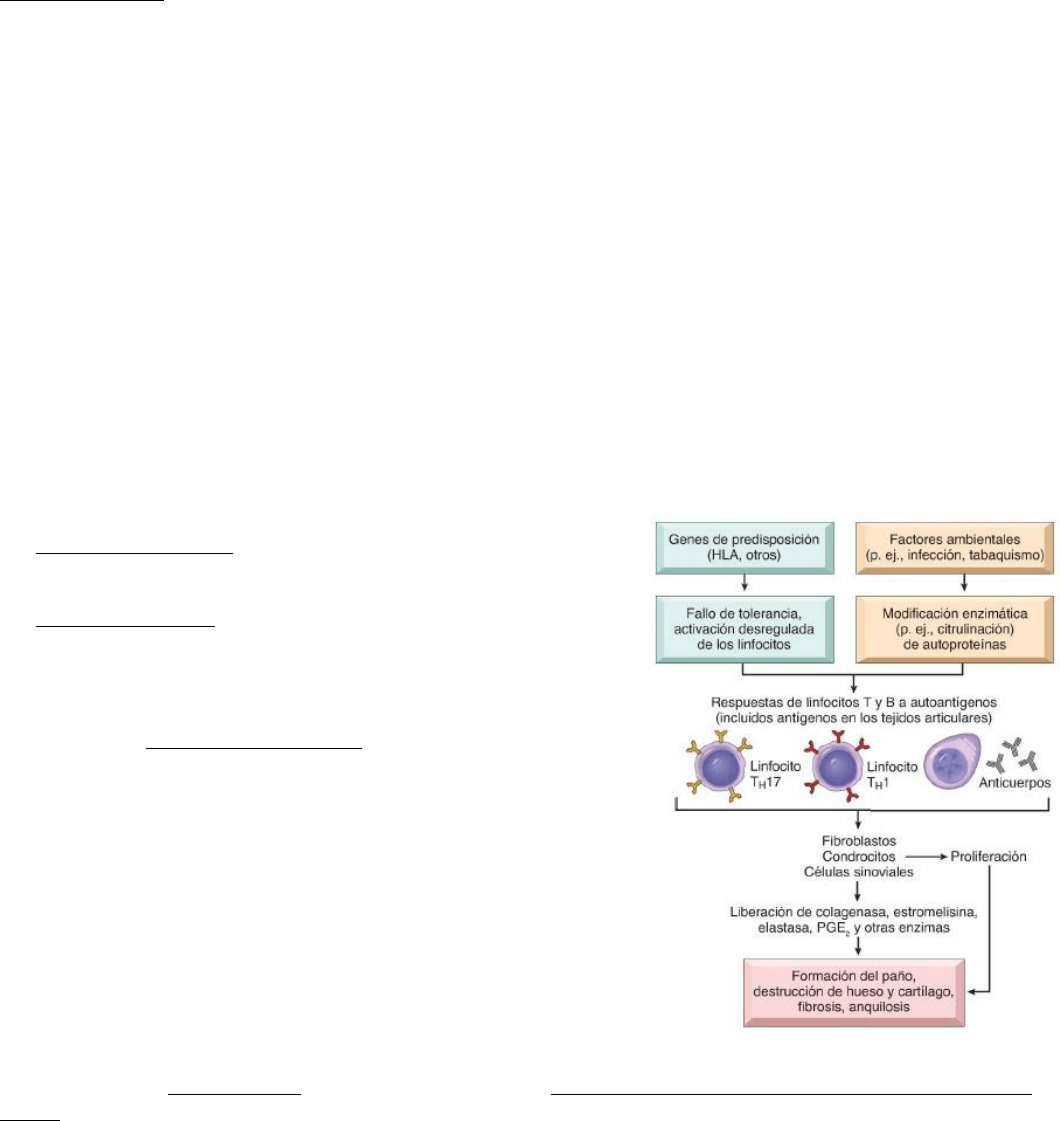
Patogenia:
-La predisposición genética y el ambiente son elementos esenciales en la aparición, progresión y cronificación de esta
enfermedad.
-Los cambios patológicos están causados por anticuerpos contra autoantígenos y por inflamación provocada por citocinas,
segregadas principalmente por los linfocitos T CD4+
Los linfocitos TCD4 pueden iniciar la rta autoinmunitaria en la AR mediante reacción contra un artriptogeno, probablemente
microbiano o autoantigeno.
Algunas de las citocinas que se detectan:
IFN-gamma: activan Mo y celulas sinoviales,
IL-7: quimiotactico
TNF e IL-1: estimulan a las celulas sinoviales residentes para que segreguen proteasas que destruyen el cartílago hialino
y
RANKL: expresado por los linfocitos T que estimulan la resorción osea.
-De estos el TNF es el que está más implicado en la patogenia de la AR.
La sinovial de la AR contiene centros germinales con folículos secundarios y numerosas células plasmáticas que producen
anticuerpos, algunos dirigidos contra autoantígenos.
Existen factores genéticos y factores ambientales.
Dentro de los genéticos: Se asocia a alelos HLA-DRB1 específicos, que
generan un fallo de la tolerancia y una activación desregulada de los
linfocitos.
Factores ambientales: una infeccion o el tabaquismo pueden generar
una modificación enzimática como una citrunilacion de autoproteinas
como las mencionadas (fibrinógeno, colágeno tipo 2, vimentina ,etc)
Muchos anticuerpos producidos en los órganos linfáticos y en la sinovial son
específicos de los péptidos citrulinados (CCP), en los que los residuos de
arginina se convierten en citrulina después de la traducción. En la AR, se
depositan en las articulaciones complejos antígeno-anticuerpo que contienen
fibrinógeno, colágeno de tipo II, a-enolasa y vimentina citrulinados. Los
anticuerpos contra estos péptidos son marcadores diagnósticos de la
enfermedad y pueden estar implicados en la lesión articular. Los datos
indican que la concentración elevada de anti-CCP, combinada con la
respuesta de los linfocitos T a las proteínas citrulinadas, contribuye a que la
enfermedad se haga crónica. Además, cerca del 80% de los pacientes tienen
anticuerpos séricos IgM o IgA que se unen a las porciones Fc de su propia IgG
(factor reumatoide).
Morfología
Articulaciones:
-se manifiesta como artritis simétrica que afecta principalmente las articulaciones pequeñas de las manos (interfalangicas) y de
los pies
-la sinovial esta edematosa, engrosada e hiperplásica, con transformaciones de su contorno liso en otro cubierto por
vellosidades frágiles y bulbosos
- Los hallazgos característicos son: 1) hiperplasia de las celulas sinoviales y proliferación de éstas 2) Infiltrados inflamatorios
densos que con forman folículos linfáticos de LT CD4+, células B, células plasmáticas, dendríticas y macrófagos; 3) aumento de
vascularización por angiogenia, 4)exudado fibrinopurulento sobre la sinovial y las superficies articulares y 5) actividad
osteoclástica en el hueso subyacente, que permite a la sinovial penetrar en el hueso y causar erosiones yuxtaarticulares, quistes
subcondrales. En conjunto los cambios mencionados producen un paño sinovial (pannus): una masa sinovial edematosa, celulas
inflamatorias, tejido de granulación y fibroblastos que crece sobre el cartílago articular y lo erosiona.
-Con el tiempo el paño sinovial se extiende entre los huesos yuxtapuestos y produce una anquilosis fibrosa, que finalmente se
osifica y produce una fusión de los huesos denominada anquilosis ósea
Piel
-Los nódulos subcutáneos reumatoides son las lesiones cutáneas más frecuentes
- Están presentes en el 25% de los casos, habitualmente con enfermedad grave y surgen en regiones de la piel sometidas a
presión, como la región cubital del antebrazo, codo, occipucio y región lumbosacra

Página 3 de 12
- Con menos frecuencia en pulmón, bazo, pericardio, miocardio, válvulas, aorta, etc
- Son firmes, indoloros a la palpación y redondos a ovales y asientan en el tejido subcutáneo de la piel
-Microscópicamente se componen de granulomas necrosante con una zona central de necrosis fibrinoide rodeada de un halo
prominente de histiocitos epitelioides.
Vasos
- La vasculitis reumatoide es una complicación grave, sobre todo si afecta órganos vitales. Similar a la PAN, excepto que no
afecta a los riñones
- Con frecuencia, segmentos de arterias pequeñas, como los vasos de los nervios y las arterias digitales, que provoca neuropatía
periférica,
- endoarteritis obliterante da ulcera y gangrena
- La venulitis leucocitoclastica produce purpura, ulceras cutáneas e infarto del lecho subungueal
Clínica
- es muy variable, comienza lentamente de modo insidioso; al principio genera malestar general, cansancio y dolor
osteomuscular generalizados (por TNF-IL-1). Luego comienza la afectación de las articulaciones. Por lo general es simétrico y
afecta a las pequeñas articulaciones. Los síntomas aparecen en las manos (metacarpofalangicas e interfalangicas) y en los pies,
seguidos por las muñecas, -- Hay inflamación de los tendones, ligamentos y los músculos esqueléticos colindantes, que produce
la desviación radial de la muñeca, desviación cubital de los dedos y anomalías en flexión-hiperextensión de los dedos
(deformidad en cuello de cisne, deformidad en ojal). El resultado son articulaciones deformadas sin estabilidad y con un arco de
movilidad mínimo o nulo
-Pueden aparecer quistes sinoviales grandes como el quiste de Baker en la región posterior de la rodilla ya que el aumento de la
presión intraarticular produce una herniación de la sinovial
-Los signos rx distintivos son: derrames articulares, osteopenia yuxtaarticular, erosiones y pinzamiento de espacio articular y
perdida del espacio articular (anquilosis)
-El diagnostico de AR se basa en 1) hallazgos radiológicos caracteristicos, 2) liquido sinovial turbio pero estéril, con poca
viscosidad, formación defectuosa del coágulo de mucina y PMN, con inclusiones, 3) combinación de factor reumatoide y de
anti-CCP
HUESO
ANOMALIAS CONGÉNITAS DE LAS CÉLULAS, LA MATRIZ Y LA ESTRUCTURA ÓSEA
Las anomalías congénitas tienen base genética y se manifiestan en etapas iniciales de la formación ósea. Las adquiridas suelen
detectarse en la edad adulta.
Disostosis: anomalía congénita por problemas localizados en la migración de las células mesenquimatosas y en la formación de
las condensaciones. Suelen afectar sólo a estructuras embriológicos concretas y están causadas por mutaciones en factores de
transcripción (genes de homeosecuencia).
Displasias: son trastornos con mutaciones en los reguladores de la organogenia ósea como las moléculas de señalización y los
componentes de la matriz que afectan globalmente a los tejidos óseos y cartilaginosos.
Malformaciones y Enfermedades causadas por defectos en las proteínas y factores de transcripción nucleares
Son infrecuentes, y comprenden el fallo de desarrollo de un hueso (ausencia congénita de una falange), la formación de huesos
adicionales (costillas o dedos supernumerarios), o la fusión de dos o más dedos (sindactilia). Están causadas por defectos en la
formación de las condensaciones mesenquimatosas y en su diferenciación en el primordio de cartílago. Están mutados los genes
de homeosecuencia y ciertas citocinas.
Displasia cleidocraneal, un trastorno autosómico dominante, provocado por mutaciones en el gen RUNX2, caracterizado por
fontanelas persistentes, cierre diferido de las suturas craneales, huesos wormianos, retraso de la erupción de los dientes
secundarios, clavículas primitivas y talla baja.
Enfermedades asociadas a defectos en las proteínas estructurales extracelulares
Enfermedades del colágeno 1 (osteogenia imperfecta)
La enfermedad de los huesos frágiles es un trastorno causado por deficiencias en la síntesis de colágeno tipo 1.
-Es el trastorno hereditario más frecuente que afecta huesos, articulaciones, ojos, orejas, etc. Se debe a mutaciones
autosómicas dominantes en los genes que codifican las cadenas 1 y 2 del colágeno. Las mutaciones que disminuyen la síntesis
de colágeno cualitativamente normal están asociadas a anomalías esqueléticas leves. Los fenotipos más graves son los que no
pueden formar la triple hélice.
Hay cuatro subtipos:

Página 4 de 12
Variante tipo II, es siempre mortal durante la gestación o en el período perinatal. Hay fragilidad ósea extrema con múltiples
fracturas en la etapa intrauterina.
Variante tipo I, esperanza de vida normal, sufren fracturas en la infancia que disminuyen tras la pubertad.
Esclerosis azules, hay una disminución del contenido de colágeno que provoca que la esclerótica sea translúcida y permita
ver parcialmente la coroides subyacente.
Perdida de la audición, relacionada con un defecto neurosensitivo y con un impedimento de la conducción por anomalías en
los huesos del oído medio e interno; las imperfecciones dentales (dientes pequeños, deformes y azul-
amarillo) por la deficiencia en la dentina.
Enfermedades asociadas a defectos en las vías metabólicas
Osteopetrosis
O enfermedad de los huesos de mármol o enfermedad de Albers-Schönberg, es un grupo de enfermedades genéticas
infrecuentes caracterizadas por resorción ósea reducida y esclerosis ósea simétrica difusa por formación o función anormal de
los osteoclastos. Los huesos son anormalmente frágiles y se fracturan con facilidad, como una barra de tiza.
PATOGENIA
- La mayoría de las mutaciones interfieren con el proceso de acidificación de la laguna de resorción del osteoclasto necesario
para disolver la hidroxiapatita cálcica del interior de la matriz. Los defectos autosómicos recesivos del gen CA2 provocan la
ausencia de la anhidrasa carbónica II que impide al osteoclasto acidificar la launa de resorción y disolver la hidroxiapatita, y
también bloquea la acidificación de la orina por las células tubulares renales.
MORFOLOGÍA
-Los huesos carecen de conducto medular y los extremos de los huesos largos son bullosos (deformidad en matraz de
Erlenmeyer) y deformes. Los agujeros neurales son pequeños y comprimen los nervios salientes. La esponja primaria, que
normalmente desaparece con el crecimiento, persiste y ocupa la cavidad medular, por no que no deja sitio para la médula
hematopoyética e impide la formación de trabéculas maduras. El hueso depositado no sufre remodelado y tiende a una
estructura reticular. Histológicamente, el número de osteoclastos puede ser normal, alto o bajo según el defecto genético
subyacente.
CLÍNICA
- La osteopetrosis maligna grave del lactante es autosómica recesiva, y se manifiesta durante la gestación o después del parto.
Hay fracturas, anemia e hidrocefalia que provocan la muerte posparto. Los que sobreviven tienen defectos en los nervios
craneales e infecciones repetidas por la insuficiencia de médula ósea que provoca hepatoesplenomegalia prominente. La forma
autosómica dominante benigna puede pasar inadvertida hasta la adolescencia o edad adulta, hay anemia y déficits leves de los
nervios craneales.
Enfermedades asociadas a una reducción de la masa ósea
Osteoporosis
Caracterizada por huesos porosos y médula ósea reducida. El trastorno puede estar localizado en cierto hueso o región, como la
osteoporosis por desuso de una extremidad, o puede afectar a todo el esqueleto como manifestación de una enfermedad ósea
metabólica. La osteoporosis generalizada puede ser primaria o secundaria.
PATOGENIA
La masa ósea máxima se alcanza al principio de la etapa adulta. Su magnitud está determinada por factores hereditarios,
actividad física, fuerza muscular, dieta y estado hormonal. Una vez alcanzada la masa ósea máxima, sobreviene un ligero déficit
en la formación de hueso con cada ciclo de resorción y formación de cada unidad multicelular básica. Por lo tanto, la pérdida de
masa ósea asociada a la edad es un fenómeno biológico normal y previsible, y afecta a ambos sexos por igual.
Hipótesis sobre la patogenia de la osteoporosis:
Cambios asociados con la edad. Los osteoblastos de las personas ancianas tienen una capacidad reducida de proliferación y
biosíntesis en comparación con los osteoblastos de los jóvenes. Las proteínas unidas a la matriz extracelular pierden su
potencia biológica con el tiempo. El resultado es una capacidad disminuida para formar hueso. Esta forma de osteoporosis
senil se clasifica como variante con recambio bajo.
Actividad física escasa aumenta la tasa de pérdida ósea, porque las fuerzas mecánicas estimulan el remodelado óseo
normal.
Factores genéticos el 60-80% de la variación en la densidad ósea está determinada genéticamente. Los principales genes
asociados son RNAKL, OPG y RANK, que codifican reguladores clave de los osteoclastos.
Estado nutricional de calcio es importante. Las mujeres adolescentes tienen tendencia a un consumo dietético de calcio
insuficiente y tienen más riesgo de sufrir osteoporosis.
Insuficiencias hormonales. La osteoporosis posmenopáusica se caracteriza por una aceleración dependiente de hormonas
de la pérdida ósea. Es probable que la reducción de la concentración de estrógenos aumente la secreción de citocinas

Página 5 de 12
inflamatorias por monocitos sanguíneos y células de la médula ósea. Hay una actividad osteoblástica compensadora, aunque
no al mismo ritmo, por lo que se produce una osteoporosis por recambio alto.
MORFOLOGÍA. La osteoporosis posmenopáusica y la senil afectan a todo el esqueleto. En la osteoporosis posmenopáusica, el
aumento de la actividad osteoclástica afecta principalmente a los huesos o regiones de los huesos con aumento del área de
superficie (cuerpos vertebrales). Las láminas esponjosas presentan perforaciones, se adelgazan y pierden sus interconexiones
con aparición de microfracturas progresivas y colapso vertebral. En la osteoporosis senil la cortical se adelgaza por resorción
subperióstica y endóstica y los sistemas haversianos se ensanchan. La cortical parece hueso esponjoso, el resto del hueso tiene
una composición normal.
CLÍNICA. Depende de los huesos afectados. Las fracturas vertebrales frecuentes en la columna torácica y lumbar producen
dolor y pueden causar una pérdida de altura considerable y distintas deformidades (lordosis lumbar y cifoescoliosis). Las
complicaciones de las fracturas del cuello femoral, pelvis o vértebras, como la embolia pulmonar y la neumonía, con
frecuentes y mortales.
Es difícil establecer un diagnóstico preciso, ya que permanece asintomática hasta que la fragilidad ósea está muy avanzada. La
prevención y el tratamiento son ejercicio, consumo adecuado de calcio y vitamina D y los fármacos que se unen al hueso e
inhiben los osteoclastos.
Enfermedades causadas por disfunción del osteoclasto
Enfermedad de Paget (osteítis deformante)
Presenta tres fases:
1. Fase osteolítica inicial
2. Fase osteoclástica-osteoblástica mixta que acaba con predominio de actividad osteoblástica
3. Fase osteoesclerótica inactiva de desgaste
El efecto neto es un aumento de la masa ósea. Sin embargo, el hueso neoformado esta desordenado y es estructuralmente.
Comienza alrededor de los 70 años.
PATOGENIA. Desconocida, y se sospecha de factores ambientales y genéticos. El riesgo de padecer la enfermedad aumenta 7
veces en familiares de primer grado. Las mutaciones en el gen SQSTM1 están presentes en 40-50% de los casos familiares y en 5-
10% de los casos esporádicos. Las mutaciones SQSTM1 aumenta la actividad de NF-kβ mediante señalización RANK, con
aumento de la actividad del osteoclasto y aumento de la propensión a la enfermedad.
Morfología
- Es un proceso local con notable variabilidad histológica con el tiempo y entre distintas localizaciones. El signo distintivo es el
patrón en mosaico del hueso laminar, es similar al rompecabezas y se debe a líneas de adhesión prominentes que ensamblan las
unidades de hueso laminar dispuestas al azar. En la fase lítica inicial, hay ondas de actividad osteoclástica y numerosas lagunas
de resorción. Los osteoclastos son anormalmente grandes y tienen muchos núcleos. Ellos persisten en la fase mixta, aunque
muchas de las superficies óseas están revestidas por osteoblastos prominentes. La MO adyacente es sustituida por tejido
conjuntivo laxo, que contiene células osteoprogenitoras y numerosos vasos sanguíneos. El hueso neoformado puede ser
reticular o laminar. A medida que el patrón en mosaico se extiende y la actividad celular decrece, el tejido fibrovascular perióseo
retrocede y es sustituido por médula ósea normal. Al final, el hueso es más grande de lo normal y está formado por trabéculas y
hueso cortical con engrosamiento tosco, pero blandos y porosos sin estabilidad estructural. Estos cambios aumenta la
vulnerabilidad del hueso a la deformación bajo presión, por lo que se fractura con facilidad.
Clínica
-La mayoría de los casos son leves y se descubre como hallazgo radiológico casual. Se afecta al esqueleto axial o la porción
proximal del fémur hasta en el 80% de los pacientes. Ningún hueso está exento pero es inusual que afecte costillas, peroné y
huesos de las manos y de los pies. Es frecuente el dolor localizado en el hueso afectado. Esta causado por microfracturas o por
proliferación ósea que comprime las raíces de nerviosas raquídeas y craneales. El aumento del tamaño del esqueleto
craneofacial puede producir una leontiasis ósea y un cráneo pesado que inclina la cabeza. El hueso pagético debilitado puede
provocar invaginación de la base de cráneo (platibasia) y compresión de las estructuras de la fosa posterior. El soporte de peso
produce arqueamiento anterior de los fémures y tibias, y distorsiona las cabezas femorales con aparición de artrosis secundaria
avanzada. Las fracturas en barra de tiza son frecuentes y afectan a los huesos largos de las extremidades inferiores. Las fracturas
por compresión de la columna vertebral provocan lesión medular espinal y cifosis. La hipervascularización del hueso pagético
calienta la piel que lo cubre, y en la enfermedad poliostótica extensa el aumento del flujo sanguíneo actúa como una fístula
arteriovenosa, que causa insuficiencia cardiaca. En el hueso pagético son frecuentes diversos tumores y lesiones
seudotumorales.

Página 6 de 12
Osteomielitis
Indica inflamación del hueso y la medula osea, siempre secundaria a una infeccion.
Se manifiesta como un foco primario de infeccion.
Osteomielitis piógena:
-ocasionada casi siempre por infecciones bacterias
-estafilococo áureas es el responsable del 85% de los casos de osteomielitis piógena.
-En la micro: se observa necrosis osea llamado secuestro oseo (hueso necrótico)
FRACTURAS
Traumáticas o no son uno de los trastornos principales de los huesos.
Se clasifican como:
completas o incompletas,
cerradas (simples) cuando no hay lesión en la piel,
abiertas cuando el foco de fractura comunica con el exterior del cuerpo,
conminutas cuando existen muchos fragmentos o
desplazadas cuando los extremos fracturados no están alineados.
-Si afecta a un hueso ya debilitado por una enfermedad subyacente como un tumor se llama fractura patológica.
-Una fractura por estrés es un que aparece lentamente tras un período de tiempo de aumento de la actividad física en el que el
hueso es sometido a nuevas fuerzas repetitivas.
El hueso es único en su capacidad de autorreparación. Puede reconstruirse por completo mediante procesos reactivos que
normalmente tienen lugar durante la embriogenia.
TUMORES Y LESIONES SEUDOTUMORALES DEL HUESO
-Son muchos más frecuentes los tumores benignos que los malignos.
-Los benignos aparecen en las 3 primeras décadas y los malignos en adultos mayores.
-Tiene predomino masculino excepto en de tumor de celulas gigantes.
-Pueden ser:
Neoplasia primarias: surgen mayormente en las extremidades inferior, sobretodo en la rodilla.
Neoplasias secundarias: Se dan más en el esqueleto axial, sobretodo en las vértebras. En adultos mayores de 45 superan a
las neoplasias primarias y provienen de próstata, pulmón, mama, riñón, aparato digestivo y tiroides.
En niños: los tumores más comunes son: Neuroblastoma, Tumor de Wilms (tumor renal), Sarcoma de Edwing y
Rabdomiosarcoma.
-Sin embargo el hueso se afecta más por neoplasia hemolinfoides (linfoma y LMC en adultos y LMA en niños)
Patogenia:
-La mayoría son idiopáticas
-Algunas se vinculan con la herencia (osteosarcoma/Sindrome de Li-Fraumeni) y el retinoblastoma.
-Otras se asientan sobre hueso enfermo.
-Puede aparecer después de tratamiento ionizante (puede aparecer entre 4-40 años)
Clasificación de neoplasias primarias
Diagnostico
-se realiza mediante trípode: clínica-radiología-anatomopatologica
1) Clínica
-Observar si estoy frente a un hueso sano o enfermo (antecedentes)
Página 7 de 12
-Edad: ya mencionado (antes de los 30 posible neoplasia 1°, si es después de los 45 años sospecha de metástasis)
-Sexo: ligero predominio masculino excepto por el de celulas gigantes
-Sintomas: dolor-tumefacción/ritmo de crecimiento (Osteosarcoma)- alteración de estado general (Edwing)- aumento de
temperatura
Localizacion
Metafisis: Osteocondroma- Osteoma Osteoide- Osteoblastoma-Osteosarcoma
Diáfisis: Sarcoma de Edwing- Displasia Fibrosa- Condrosarcoma
Epífisis: Tumor de Celulas Gigantes- Condroblastoma
2) Radiología
-Mirar desde afuera hacia adentro
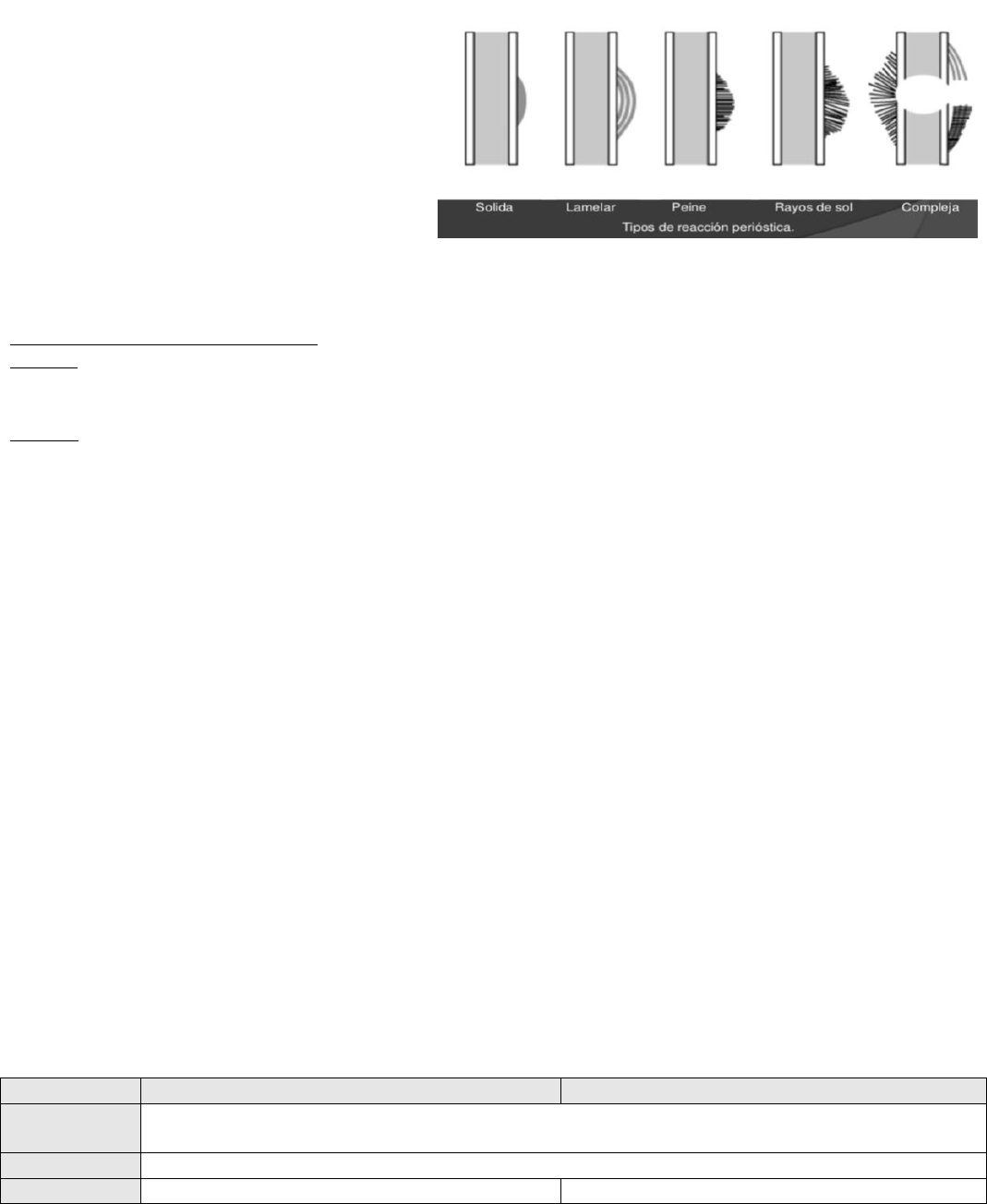
-Observar la Reacción Perióstica:
solida: neoplasias benignas
Lamelar-Peine-Rayos de sol-Compleja:
neoplasias malignas
RMN: para definir compromiso en partes blandas
Gammagrafía: define áreas de captación
3) Anatomopatologica
-Tipo histológico del tumor
-Grado Histológico
-Grado de rta al tto adyuvante
Neoplasias antes de los 20 años
Benignas
Osteocondroma
Osteoma Osteoide/ Osteoblastoma
Malignos
Sarcoma de Edwing
Osteosarcoma
Osteocondroma (también denominado exostosis, sale hacia fuera)
-Es el tumor más frecuente benigno alrededor del 85% son solitarios, el resto puede formar del Sme de exostosis hereditaria
múltiple.
-Afecta más a los hombres
-Se localiza en la Metafisis, nace en el cartílago del crecimiento se cierra la placa y no crece más.
-Es un tumor con una cubierta de cartílago unido al hueso subyacente por un pedúnculo oseo.
-Los solitarios se Dx en la adolescencia y principio de la edad adulta, los Osteocondroma múltiples son aparentes en la infancia.
-Aparecen solo en huesos de origen endocondral y se originan en la Metafisis cerca del cartílago del crecimiento, sobretodo en
la rodilla. Puede aparecer en escapula, costilla y pelvis, en estos lugares suele ser sésiles.
Patogenia: mutación del gen EXT-1 y EXT-2, gen que codifica glucosaminoglucanos y heparan sulfato que provocan crecimiento
excesivo de cartílago y oseo.
Morfología
Son sésiles o pediculados. La cubierta está formada por cartílago hialino benigno y cubierto por pericondrio. El cartílago muestra
un crecimiento desorganizado y sufre osificación endocondral, de forma que el hueso nuevo forma la porción interna de la
cabeza y el pediculo. La cortical del hueso nuevo se fusiona con la del hueso de donde nace, de modo que se conectan con la
medula osea.
Clínica
-Deja de crecer habitualmente en el momento del cierre del cartílago de crecimiento.
-Algunos se curan con extirpación simple. En la exostosis hereditaria múltiple puede progresar a condrosarcoma,
-Son masas de crecimiento lento e indoloros, excepto si se comprime un nervio o fractura un pediculo.
Osteoma Osteoide/ Osteoblastoma
Osteoma Osteoide
Osteoblastoma
Caracteristicas
Generales
Son tumores productores de hueso, con características histológicas idénticas.
Edad
Adolescencia y tercera década
Tamaño
< 2cm
>2cm

Página 8 de 12
Lugar de origen
Cortical
Medular
Localizacion
Fémur y tibia
Columnar (laminas y pedículos de las vértebras)
Rx
Signo de la escarapela: Consiste en una radio
trasparencia redonda con mineralización central y su
parte más externa rodeada por un engrosamiento
cortical (hiperostosis)
Morfología
Son masas redondas de tejido pardo hemorrágico, bien delimitadas que contiene trabéculas de hueso
reticular intercomunicada de manera aleatorias, revestidas por osteoclastos prominentes.
El estroma, es decir los espacios intertrabeculares contienen tejido conectivo laxo con numerosos capilares.
Sintomas
Dolor nocturno intenso, se produce por el aumento
de la PGE2 producida por los osteoblastos
proliferantes que genera vasodilatación en reposo y
ocasiona dolor, también duele cuando ingiere
bebidas alcohólicas, este dolor mejora con Ac.
Acetilsalicílico (aspirina)
Dolor de larga evolución que no mejora con Ac.
Acetilsalicílico y no produce reacción osea
considerable.
Tratamiento
Se trata con ablación por radiofrecuencia
Se trata mediante legrado y extirpación en bloque.
Osteosarcoma
-es un tumor maligno en el que las células cancerosas producen matriz ósea.
-Es el tumor maligno primario más frecuente del hueso, aparte del mieloma y linfoma
-Se da en menores de 20 años (75%), puede surgir en adultos mayores que presentan condiciones que predisponen al
osteosarcoma, enf. de Paget y radioterapia.
-Son tumores localmente agresivos y metastatizante.
-Localizacion metafisaria en rodilla (60%), cadera, hombro, maxilar.
Son masas dolorosas que aumentan de tamaño progresivamente a veces el primer síntoma es la fractura.
Rx: se observa una masa mixta y blastica, grande y destructiva, con bordes infiltrantes. El tumor rompe la cortical y levanta el
periostico provocando la formación de hueso periostico reactivo. La sombra triangular entre la cortical y los extremos
levantados del periostio se denomina radiográficamente triangulo de Codman (periostio en rayos de sol o peine), indicador de
agresividad.
Patogenia: presentan anomalías congénitas adquiridas, ninguna especifica del tumor, hay mutaciones muy frecuentes que
intervienen en la función de los genes RB (del retinoblastoma) y del p53 (pacientes con síndrome de Li Fraumeni desarrollan
este tumor)
Morfología:
Macro: son tumores voluminosos de color gris blanco que contienen zonas de hemorragia y generación quística, destruye la
cortical circundante y producen masas de partes blandas. Se disemina por la cavidad medular, infiltran y sustituyen la M.osea.
Micro: Posee un patrón en encaje del hueso neoplásico producido por celulas tumores malignas anaplasicas, la formación de
hueso por las celulas tumorales es diagnostica.
Clínica
-Son masas dolorosas de crecimiento progresivo que originan fracturas patológicas.
-Es un tumor osteoblastico, solitario, endomedular, poco diferenciado y agresivo.
-Se diseminan por via hematogena a pulmones.
-El pronóstico de los pacientes con metástasis es pésimo.
-Se diagnostica en momentos avanzados.
Sarcoma de Ewing/tumor neuroectodérmico primitivo (PTEN)
-Es un tumor oseo maligno caracterizados por célula redonda malignos primarios del hueso sin diferenciación.
-El sarcoma de Ewing y el de PENET se han unificado en una categoría llamada, tumores de la familia del sarcoma de Ewing.
-Es el 2° grupo más frecuente de sarcomas en la infancia, el primero es el osteosarcoma.
-Se mas en niños menores de 20 años.
-El tumor de Ewing se localiza en la diáfisis sobretodo en el fémur y la pelvis.
RX: muestra tumor lítico destructivo con bordes infiltrantes y extensión a las partes blandas circundantes. La reacción periostico
característica produce capas de hueso reactivo en forma de capas de cebolla, (catafila, Lamelar)
Patogenia:
translocación (11:22); producen factores de transcripción quiméricos que alteran la expresión de un grupo de genes diana, que
provoca proliferación y supervivencia anormales.
Morfología:
-Son masas grandes blandas de color pardo blanco, que crecen en la cavidad medular pero invaden la cavidad medular, cortical,

Página 9 de 12
el periostio y las partes blandas. Contiene zonas de hemorragia y necrosis.
-Cuando el tumor de PNET aparece en los 15 días de vida y ocupa todo el tórax se llama tumor de Asking
-La presencia de rosetas de Homer-Wright (células tumorales dispuestas en círculo alrededor de un espacio fibrilar central) es
indicativa de diferenciación neural.
Clínica
-Son masas dolorosas que aumentan de tamaño y la región afectada presenta con frecuencia dolor a la palpación, aumento de la
temperatura y tumefacción. Algunas personas pueden tener signos sistémicos (fiebre, leucocitosis, aumento de VSG)
IHQ: Marcador CD99
Neoplasias después de los 45 años
1° - Metástasis
2° - Condrosarcoma
Metástasis:
-En los adultos neoplasias malignas primarias de próstata, pulmón, mama, riñón, tiroides, tubo digestivo y piel.
-En los niños tienen su origen en un neuroblastoma, tumor de Wilms, osteosarcoma, sarcoma de Ewing y rabdomiosarcoma.
-La mayoría de las metástasis son multifocales y afectan a la columna vertebral pelvis, costilla cráneo y esternón.
-La rx puede ser lítica (destrucción del hueso), blastica (formadora de hueso) o mixta.
Condrosarcoma
-Tumor maligno productor de cartílago
- Puede ser intramedular (centrales) o yuxtacortical (periféricos)
-Hay variantes: convencional (hialina y/o mixoide, representa el 90%), célula clara, desdiferenciados y mesenquimatosa.
- Afecta a personas de 40 años o más
-Afecta más a hombres
-Localizacion: esqueleto axial (pelvis-hombros-costillas). No afecta región distal de las extremidades a diferencia del encondroma
-Rx: matriz calcificada se observa como focos de densidad confluyente, se ven como densidades flotantes
-La variante de células claras se origina de la epífisis de los huesos largos (fémur)
-Los condrosarcomas que aparecen en el Sindrome de Encondromatosis múltiple (Enfermedad de Ollier, se manifiesta como
masas de gran tamaño en los dedos de las manos) tienen mutaciones en los genes EXT
Se relaciona con la exostosis hereditaria múltiple.
-Tanto los condrosarcomas relacionados con la condromatosis como los esporádicos presentan mutaciones IDH1-IDH2
Morfología
-Los convencionales son tumores voluminosos formado por nódulos el cartílago traslucidos y la matriz es gelatinosa, es habitual
la presencia de calcificaciones punteadas.
-EL tumor se disemina a través de la cortical al musculo o la grasa circundante.
-El 10% de los condrosarcomas convencionales de bajo grado tienen un segundo componente de alto grado, con una morfología
de sarcoma poco diferenciado, que define los condrosarcomas desdiferenciados.
Hay condrocitos anaplasicos en una matriz de cartílago hialino (en un condrosarcoma de grado 3)
-El rasgo característico del condrosarcoma de células claras son las láminas de condrocitos neoplásicos malignos, numerosas
células gigantes y formación intralesiva de hueso reactivo.
-El condrosarcoma mesenquimatoso está formado por islotes de cartílago hialino diferenciado, rodeados de láminas de células
redondas pequeñas
Clínica:
son masas dolorosas de aumento progresivo.
-La mayoría de los condrosarcomas son tumores de grado 1, con alta supervivencia a los 5 años.
-Se diseminan por via hematogena al pulmón.
-Los mesenquimatosos y los desdiferenciados son más agresivos, se trata con cirugía y quimioterapia, en cambio los de celulas
claras son mucho menos agresivos.
-hay correlación directa entre el grado y la conducta biológica del tumor.
------------------------------Estos tumores que vienen no entran en la clasificación del teórico----------------------------------------------------
-----
Tumores de células gigantes
-Recibe este nombre porque en la histología predomina las celulas gigantes multinucleadas de tipo osteoclasto, que son las
causa del sinónimo osteoclastoma.
-Es un tumor benigno relativamente infrecuente, aunque localmente agresivo.
-Suele afectar entre los 20-50.

Página 10 de 12
Patogenia
-Se compone mayormente por osteoclastos y sus precursores, las celulas neoplásicas expresan una concentración elevada de
RANK-L, el resultado es una resorción de matriz localizada muy destructiva.
La mayoría son solitarios.
Morfología
-La mayoría destruye la cortical subyacente produciendo una masa abombada de partes blandas delimitada por una cubierta de
huesos.
-Son masas grandes pardo-rojizas, que a menudo presentan degeneración quística.
-Formados por células mononucleares ovales (celulas gigantes de tipo osteoclasto) con 100 nucleos o más.
-Otros signos son necrosis, hemorragia, depósito de hemosiderina
-Las celulas no sintetizan hueso ni cartílago aunque pueden estar rodeadas en la periferia por hueso
Clínica
- Localizacion: Se originan en la epífisis, la mayoría de la rodilla
-La mayoría son solitarios aunque pueden ser multicentricos
-Produce sintomas parecidos a una artritis, ya que se localizan cercanos a las articulación
-Tienen gran tasa de recidivas y se tratan con legrado
-Su conducta biológica es impredecible
Condroblastoma
Tumor benigno infrecuente en pacientes jóvenes adolescentes mayormente hombres. La mayoría asientan en la rodilla y con
menos frecuencia en pelvis y en costillas de pacientes mayoras. Tiene una predilección muy marcada por la epífisis y apófisis.
Morfología: formado por láminas de condroblastos poliédricos compactos con limites citoplásmico bien delimitados, con
frecuente actividad mitótica y necrosis. Células tumorales rodeadas de escaza matriz hialina que se deposita en configuración de
encaje. Los nódulos bien formados son infrecuentes de modo característico. Cuando se calcifica la matriz, produce un patrón de
mineralización característico “en malla de gallinero”. Dispersas hay células gigantes (osteoclastos anaplasicos). Puede presentar
degeneración quística hemorrágica prominente. Suelen ser dolorosos y pueden provocar derrame y limitación de la movilidad
articular. La recidiva tras el legrado es infrecuente.
Fibroma condromixoide
Es el menos frecuente y puede confundirse con un sarcoma. Afecta a adolescentes y a personas en la 3ra década, con
predominio masculino. Surgen en la metáfisis de huesos tubulares largos pudiendo afectar cualquier hueso.
Morfología: entre 3 y 8cm, bien delimitados, sólidos y pardo gris brillantes. Hay nódulos de cartílago hialino poco formado y de
tejido mixoide delimitados por tabiques fibrosos. Las zonas con más celularidad se | en la periferia de los nódulos. En las
regiones cartilaginosas, las células tumorales se sitúan en lagunas. En las zonas mixoides, las células son estrelladas y sus finas
proyecciones celulares se extienden por la matriz extracelular mucinosa, acercándose o contactando con las células colindantes.
Clínica: suelen referir un dolor sordo continuo localizado, las Rx muestran una radio transparencia localizada excéntrica bien
delimitada, por la existencia de un halo de esclerosis el tratamiento es el legrado simple con riesgo de recidiva pero sin riesgo de
transformación maligna ni metástasis.
Lesiones que simulan neoplasias primarias
Displasia fibrosa (desorganización del hueso y/o cartílago)
-Es un tumor benigno vinculado a una detención localizada del desarrollo.
-Están presentes todos los componentes de hueso normal, pero no se diferencian las estructuras maduras.
-Aparecen durante el crecimiento y desarrollo del esqueleto y tienen 3 patrones distintivos:
1. Monostotica: afectación de un solo hueso,
2. Poliostótica: afectación de múltiples huesos
3. Sindrome de Mazabraud: displasia fibroso poliostótica y mixoma de partes blandas.
4. Sindrome de McCune-Albright: enfermedad poliostótica asociada a pigmentación cutánea con manchas café con leche
y anomalías endocrinas.
Patogenia:
Mutación somática del gen GNAS-1
Morfología:
-lesiones circunscriptas, intramedulares y de tamaño muy diverso; las más grandes expanden y distorsionan el hueso.
-El tejido lesivo es granular pardo-blanquecino y está formado por trabéculas curvilíneas rodeadas de una proliferación
fibroblasticos. -También hay nódulos de cartílago hialino desorganizado; otros hallazgos frecuentes son la degeneración quística,
hemorragia y macrófagos espumosos. La evolución es variable y depende de la extensión de la afectación, o si tiene una
localización estratégica.
Página 11 de 12
Clínica
-La de tipo monostotica afecta por igual a niños y niñas al inicio de la adolescencia y deja de crecer al cerrarse el cartílago de
crecimiento. Los huesos más afectados son fémur, costilla, tibia, maxilar y bóveda craneal. Puede producir dolor fractura y
diferencia de longitud en las extremidades. La Rx muestra aspecto característico de vidrio esmerilado/deslustrado y sus bordes
bien definidos. Las lesiones sintomáticas curan mediante legrado.
-La de tipo poliostótica se manifiesta antes que la monostotica y causa problemas en la etapa adulta.
Los huesos más afectados son fémur, cráneo, tibia, humero, costilla, etc se asocia a enfermedad progresiva, las personas
La enfermedad tiende a afectar la cintura pélvica y escapular causando una enfermedad deformante, incapacitante que
ocasiona fractura. Puede transformarse a un sarcoma.
PARTES BLANDAS
-Se denomina partes blandas a todo derivado del mesénquima es decir los músculos esqueléticos y lisos, tejido adiposo y tejido
conectivo.
Tumores del tejido adiposo
Lipoma (tumor benigno de grasa)
Liposarcoma
C.Generales
-Es el tumor más frecuente de partes blandas en
adultos
-Se clasifica en lipoma convencional, fibrolipoma,
angiolipoma y mielolipoma
Es uno de los sarcomas más frecuente de la adultez
-Se localiza
-Patogenia: gen CHOP translocación 12:16
Edad
Adultos 40-60
Localizacion
Se localiza en el tejido subcutáneo de la región
proximal de extremidades y el tronco.
Se localiza en partes profundas cercano a las extremidades y
retroperitoneo.
Macro
Tipo convencional (más frecuente): es una masa bien
encapsulada de adipocitos maduros.
Se dividen histológicamente en 3 tipos:
1) liposarcoma bien diferenciado: contiene adipocitos con celulas
fusiformes atípicas dispersa
2) Mixoide: contiene abundante MEC basofila, y red capilar extensa
en la que hay dispersos lipoblastos (es una célula grande con nucleo
multilobulado)
3) Pleomorfo: compuesto por celulas anaplasicas y lipoblastos. Es el
que tiene peor pronostico
Micro
Se compone tejido adiposo maduro.
Clínica
Son blandos móviles e indoloros (excepto el
angiolipoma que duele)
-curan por extirpación
Todos los tipos recidivan localmente
-La variante bien diferenciada es inactiva a diferencia del tipo de
celulas redondas (mixoide) es intermedio. Mientras que la variante
pleomorfa es agresiva y metastatizante.
-Se debe indicar el grado para evaluar el pronóstico y el tratamiento.
Se indica mediante diferenciación, la actividad mitótica y presencia
de necrosis.
Tumores de musculo esquelético
Rabdomiosarcoma
Es una neoplasia maligna de extirpe mesenquimatosa con diferenciación a musculo esquelético
El Rabdomiosarcoma (alveolar y embrionario) es el sarcoma de partes blandas más frecuente que aparece en la infancia y
adolescencia (menores de 20 años)
Hay 3 tipos:
Embrionario, Alveolar, Pleomorfo (predomina en los adultos)
-En las formas infantiles se localizan en los senos paranasales en la cabeza, el cuello y aparato genitourinario.
Patogenia: hay mutación en el PAX
3
y PAX
7
con el factor de crecimiento FOX
1
Morfología:
Rabdomiosarcoma embrionario: es una masa blanda blanquecina. Las celulas tumorales forman capas de celulas
fusiformes en un estroma mixoide. Las celulas son rabdomioblastos alargados (en tirante), que puede tener el nucleos
alargados llamados nucleo en renacuajo.
-Tiene variantes: 1)sarcoma botroideo (tiene mejor pronóstico que todos) se localiza en las paredes de estructuras
huecas revestidas por mucosas (nasofaringe, vagina, vejiga urinaria)
Página 12 de 12
En la región donde los tumores contactan con los órganos forman una zonas submucosa de hipercelularidad llamada
capa cambial
Rabdomiosarcoma alveolar: esta atravesado por una red de tabiques fibrosos que separan a las celulas en cúmulos o
conglomerados, con cierto parecido con los alveolos pulmonares.
Rabdomiosarcoma pleomorfo: se caracteriza por numerosas celulas tumorales eosinófilas anómalas grandes, algunas
multinucleadas.
Clínica:
-El tipo histológico y la localización del tumor influyen en la supervivencia.
El botroide tiene mejor pronóstico mientras que el pleomorfo es mortal.

23-Patologia-osteoarticular-y-de-partes-blandas.pdf
 Estamos procesando este archivo...
Estamos procesando este archivo...
 Lamentablemente la previsualización de este archivo no está disponible. De todas maneras puedes descargarlo y ver si te es útil.
Lamentablemente la previsualización de este archivo no está disponible. De todas maneras puedes descargarlo y ver si te es útil.
Descargar
Estamos procesando este archivo...
Lamentablemente la previsualización de este archivo no está disponible. De todas maneras puedes descargarlo y ver si te es útil.